| Arginine and proline metabolism | 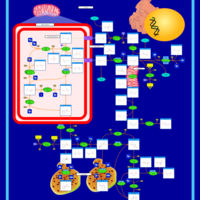 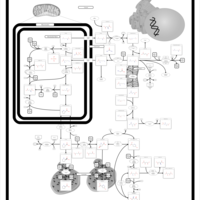 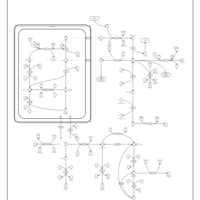 |
| Citrullinemia Type I | 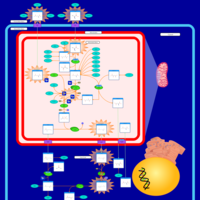 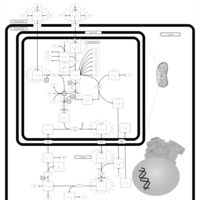 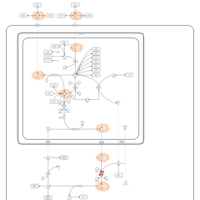 |
| Carbamoyl Phosphate Synthetase Deficiency | 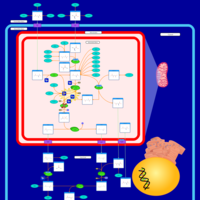 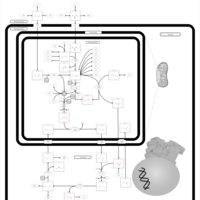 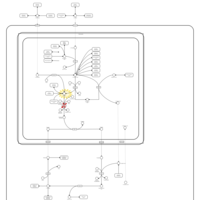 |
| Argininosuccinic Aciduria |   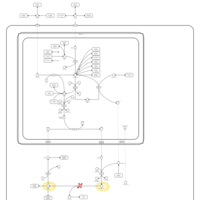 |
| D-Arginine and D-Ornithine Metabolism |  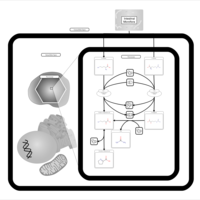 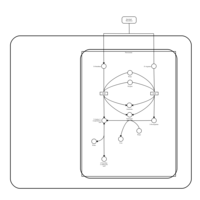 |
| Urea Cycle | 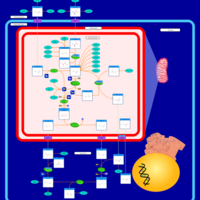 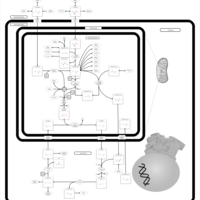 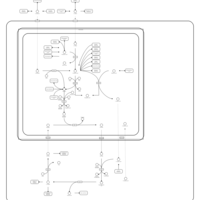 |
| Chlorothiazide Action Pathway | 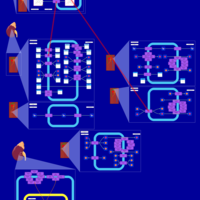 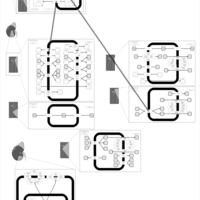 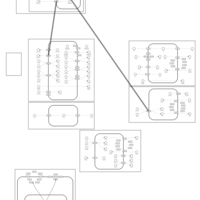 |
| Polythiazide Action Pathway |  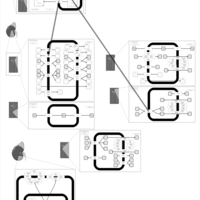  |
| Methyclothiazide Action Pathway | 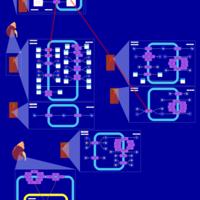 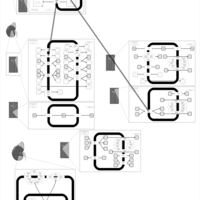 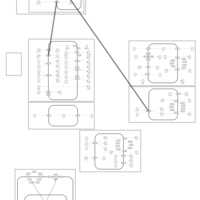 |
| Bumetanide Action Pathway | 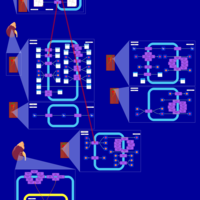   |
| Bendroflumethiazide Action Pathway | 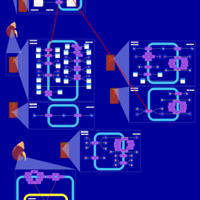 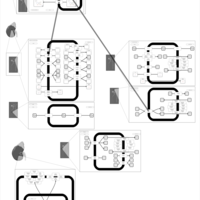 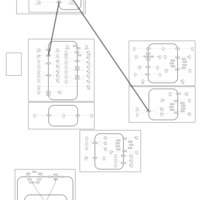 |
| Quinethazone Action Pathway | 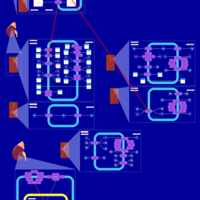 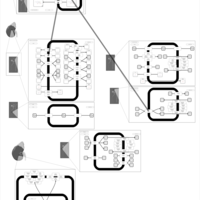 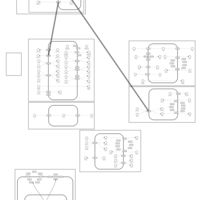 |
| Ethacrynic Acid Action Pathway | 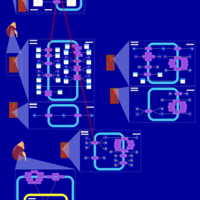 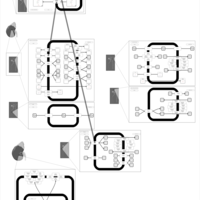 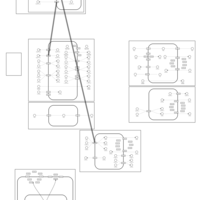 |
| Hydrochlorothiazide Action Pathway | 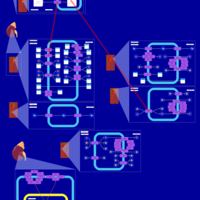 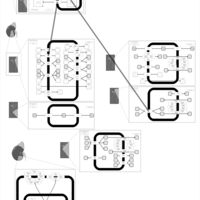 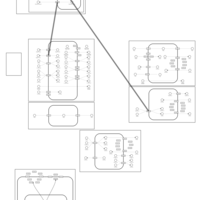 |
| Cyclothiazide Action Pathway |  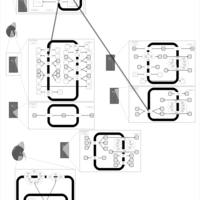 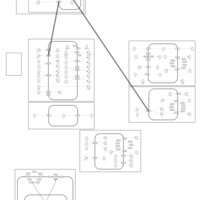 |
| Metolazone Action Pathway | 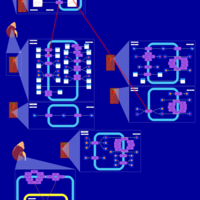  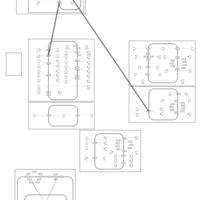 |
| Hydroflumethiazide Action Pathway | 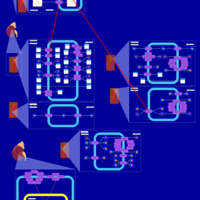 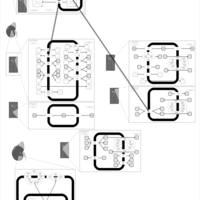 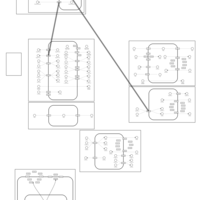 |
| Indapamide Action Pathway | 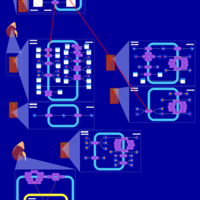 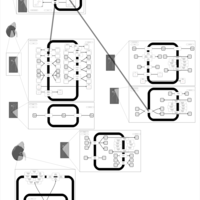 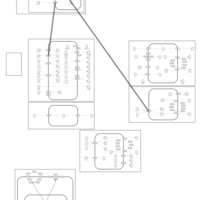 |
| Furosemide Action Pathway | 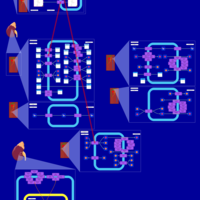 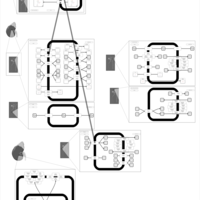 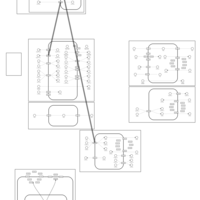 |
| Torsemide Action Pathway | 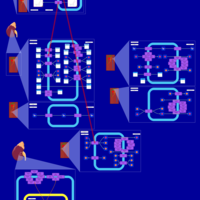   |
| Trichlormethiazide Action Pathway | 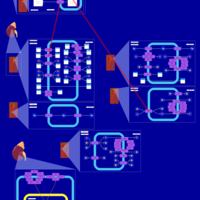 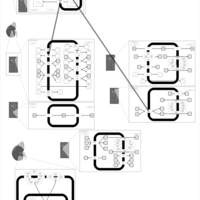 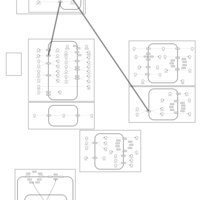 |
| Chlorthalidone Action Pathway | 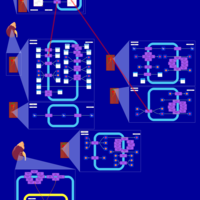 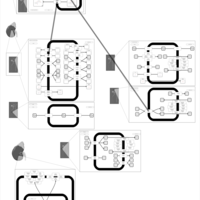 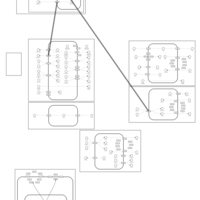 |
| Triamterene Action Pathway | 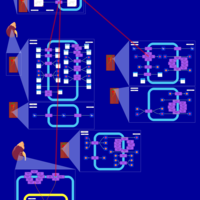 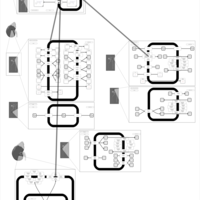  |
| Amiloride Action Pathway | 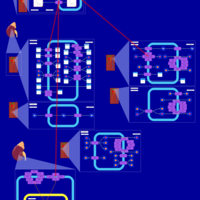 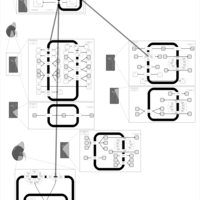 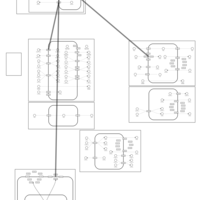 |
| Spironolactone Action Pathway |   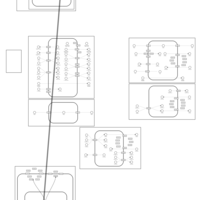 |
| Eplerenone Action Pathway | 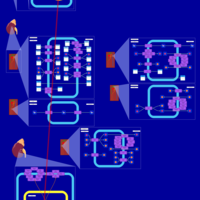 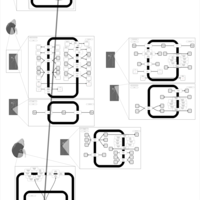 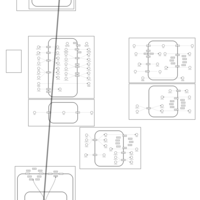 |
| Glucose Transporter Defect (SGLT2) | 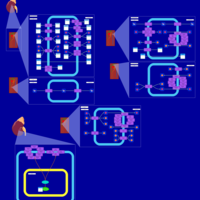   |
| Guanidinoacetate Methyltransferase Deficiency (GAMT Deficiency) |    |
| Hartnup Disorder |    |
| Iminoglycinuria | 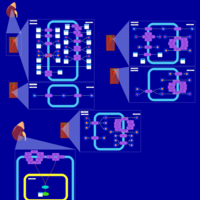 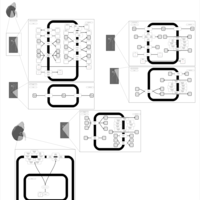 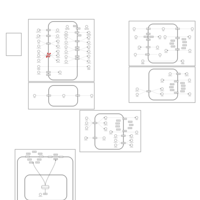 |
| Lysinuric Protein Intolerance |  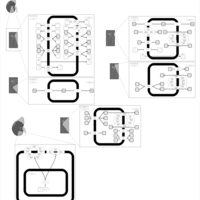  |
| Ornithine Transcarbamylase Deficiency (OTC Deficiency) | 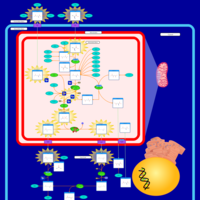 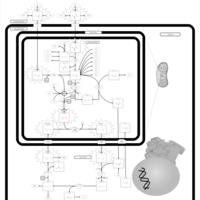 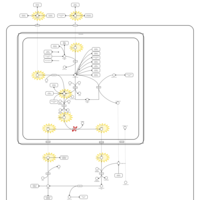 |
| Prolidase Deficiency (PD) | 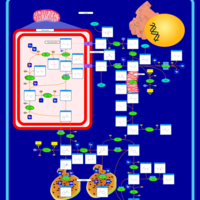 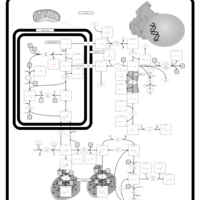 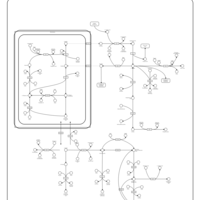 |
| Prolinemia Type II |   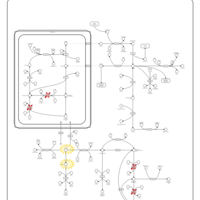 |
| Argininemia | 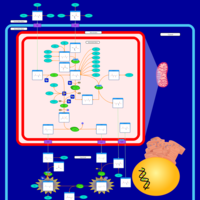  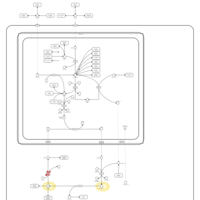 |
| Hyperprolinemia Type II | 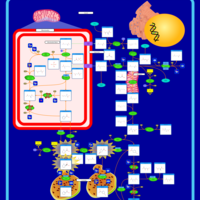 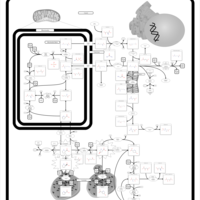 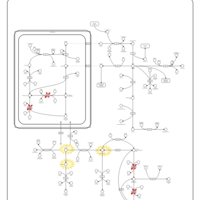 |
| Hyperprolinemia Type I | 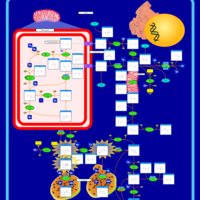 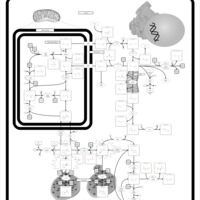 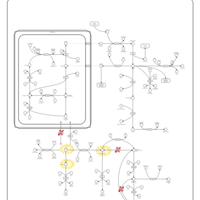 |
| Arginine: Glycine Amidinotransferase Deficiency (AGAT Deficiency) | 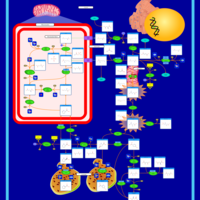 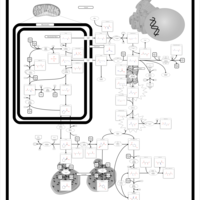 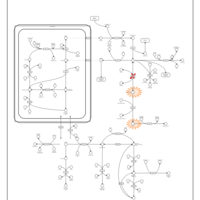 |
| Ornithine Aminotransferase Deficiency (OAT Deficiency) |    |
| Kidney Function | 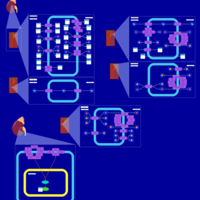 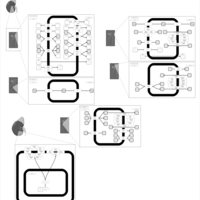  |
| Creatine deficiency, guanidinoacetate methyltransferase deficiency |    |
| Hyperornithinemia with gyrate atrophy (HOGA) |    |
| Hyperornithinemia-hyperammonemia-homocitrullinuria [HHH-syndrome] |    |
| L-arginine:glycine amidinotransferase deficiency |    |
| Blue diaper syndrome |    |
| Lysinuric protein intolerance (LPI) |    |
| Cystinuria |    |
