| Alanine, aspartate and glutamate metabolism | 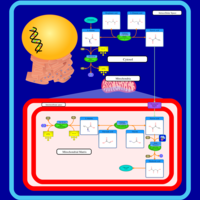   |
| Pyruvate metabolism |    |
| Tyrosine metabolism |  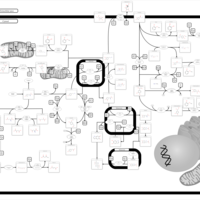 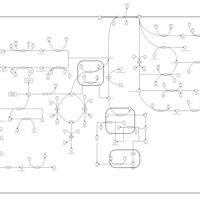 |
| Arginine and proline metabolism | 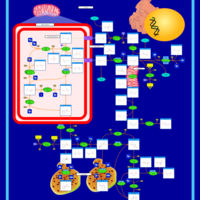 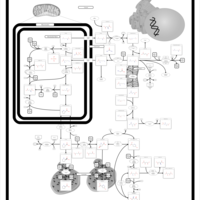 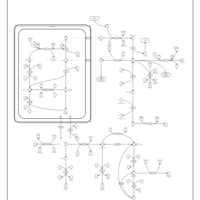 |
| Citrullinemia Type I | 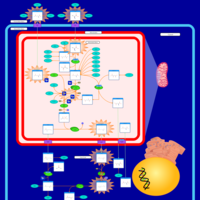 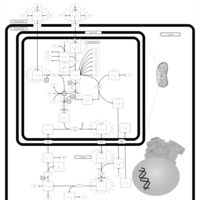 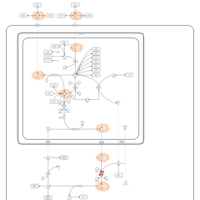 |
| Carbamoyl Phosphate Synthetase Deficiency | 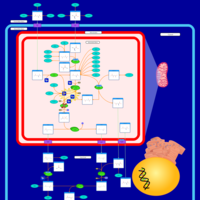 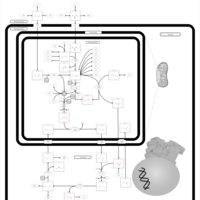 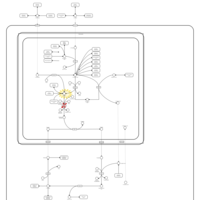 |
| Argininosuccinic Aciduria | 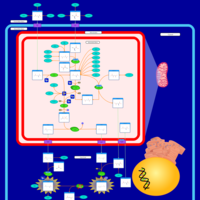  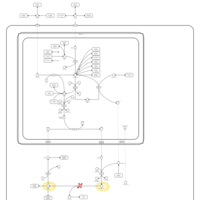 |
| Citric Acid Cycle | 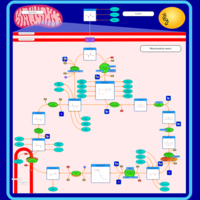 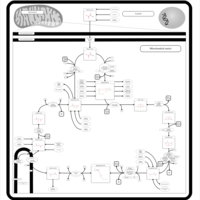 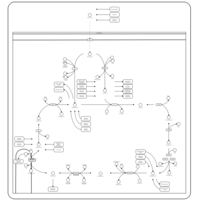 |
| Urea Cycle | 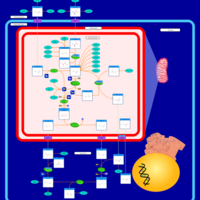 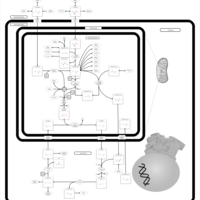 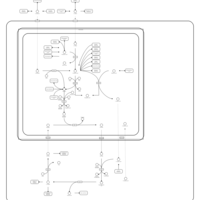 |
| Aspartate Metabolism | 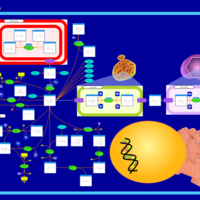 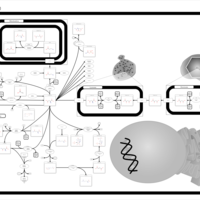 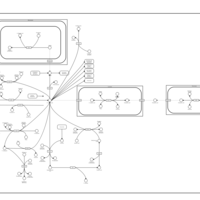 |
| Glutamate Metabolism | 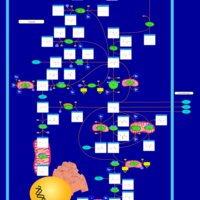 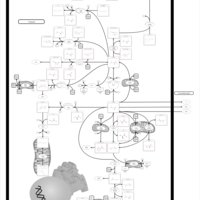 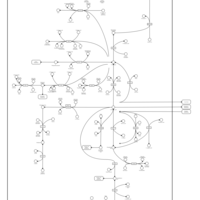 |
| Gluconeogenesis | 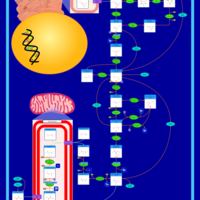 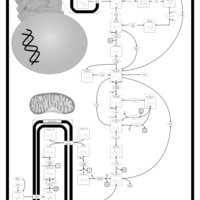 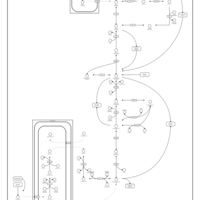 |
| Malate-Aspartate Shuttle |  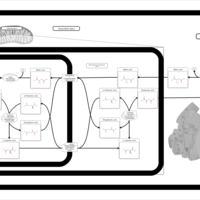 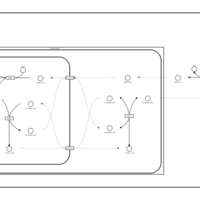 |
| 2-Hydroxyglutric Aciduria (D And L Form) | 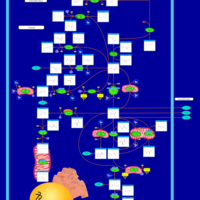 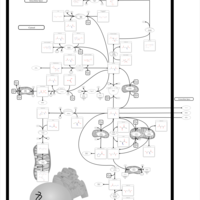 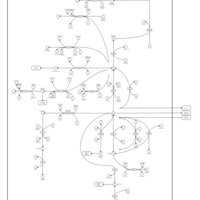 |
| Alkaptonuria | 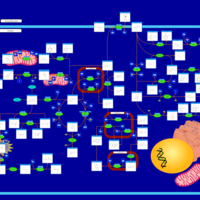 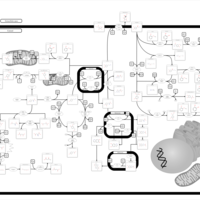  |
| Canavan Disease | 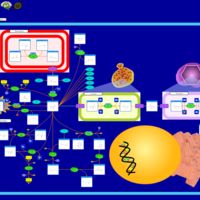 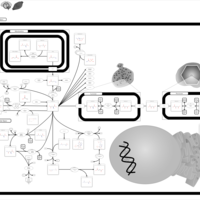  |
| Guanidinoacetate Methyltransferase Deficiency (GAMT Deficiency) |    |
| Hawkinsinuria | 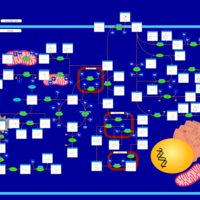  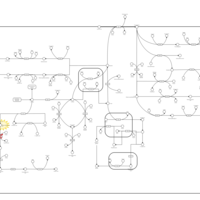 |
| Hypoacetylaspartia | 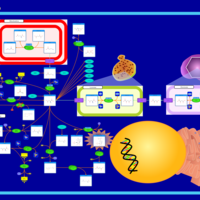 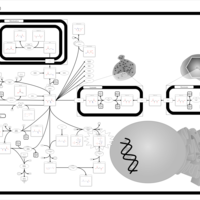  |
| Leigh Syndrome | 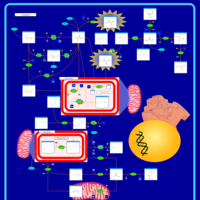 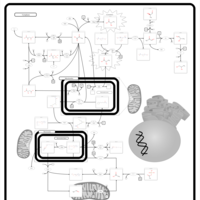 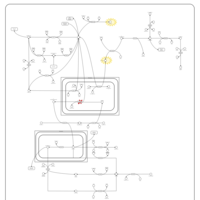 |
| Ornithine Transcarbamylase Deficiency (OTC Deficiency) | 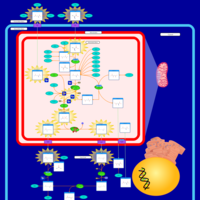 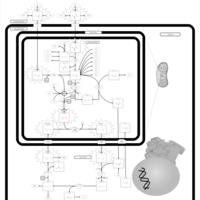 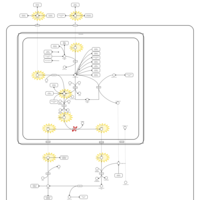 |
| Prolidase Deficiency (PD) | 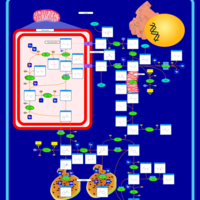 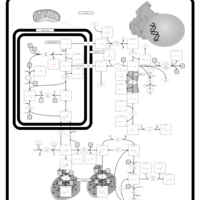  |
| Prolinemia Type II |   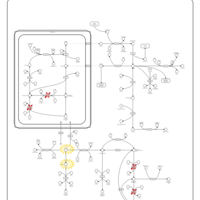 |
| Pyruvate Dehydrogenase Complex Deficiency | 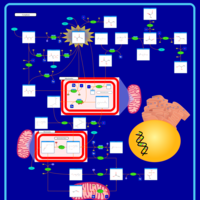 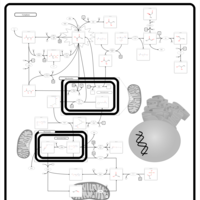 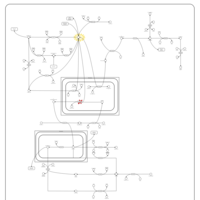 |
| Tyrosinemia Type I | 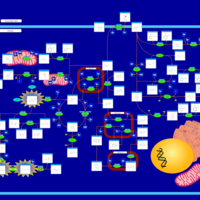 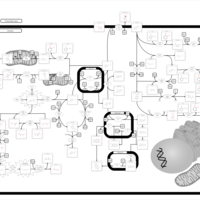 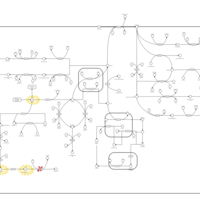 |
| 4-Hydroxybutyric Aciduria/Succinic Semialdehyde Dehydrogenase Deficiency | 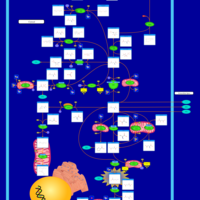 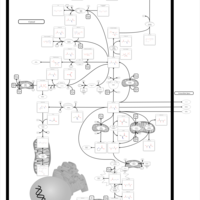 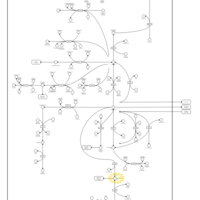 |
| Lactic Acidemia | 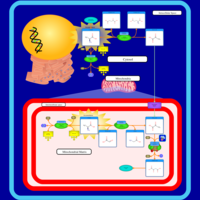 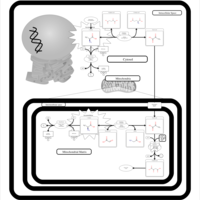 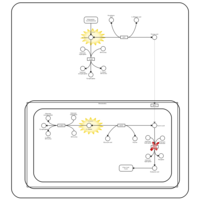 |
| Pyruvate Decarboxylase E1 Component Deficiency (PDHE1 Deficiency) | 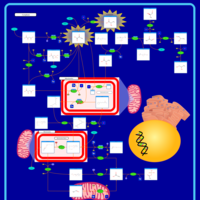 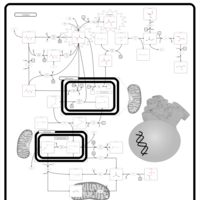 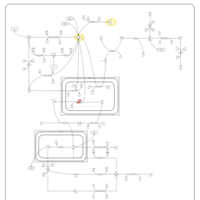 |
| Hyperinsulinism-Hyperammonemia Syndrome | 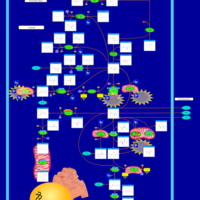 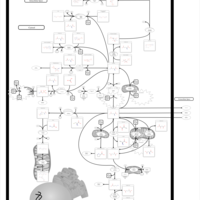 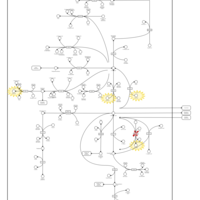 |
| Pyruvate Carboxylase Deficiency | 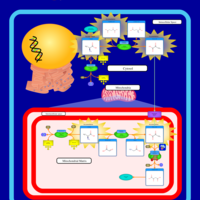 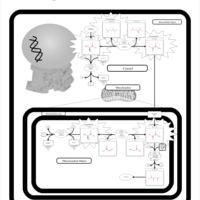 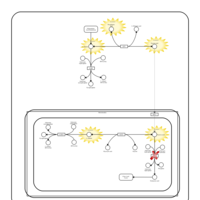 |
| Primary Hyperoxaluria Type I | 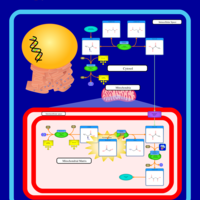 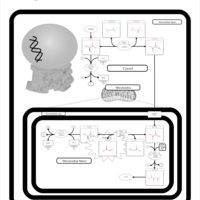 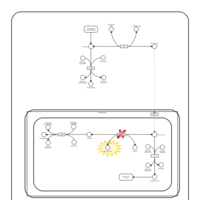 |
| Argininemia | 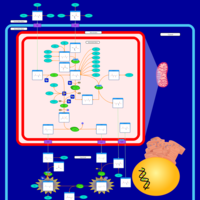  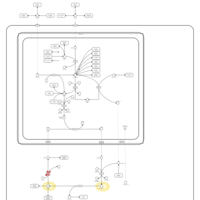 |
| Hyperprolinemia Type II | 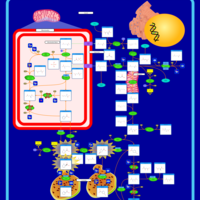 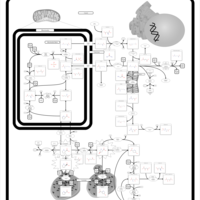 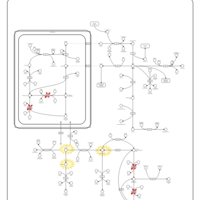 |
| Hyperprolinemia Type I | 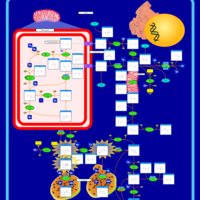 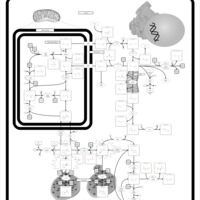 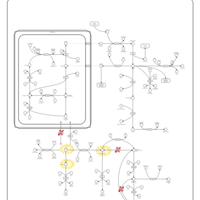 |
| Arginine: Glycine Amidinotransferase Deficiency (AGAT Deficiency) | 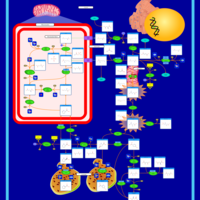 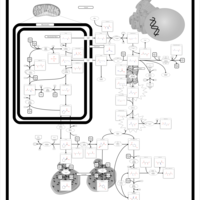  |
| Ornithine Aminotransferase Deficiency (OAT Deficiency) |    |
| Glycogen Storage Disease Type 1A (GSD1A) or Von Gierke Disease | 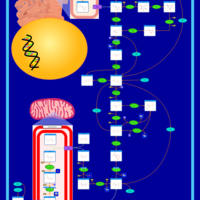 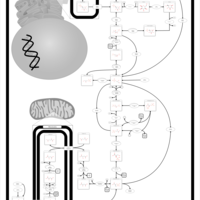 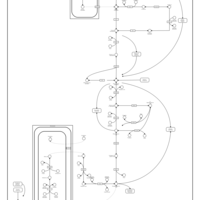 |
| Homocarnosinosis | 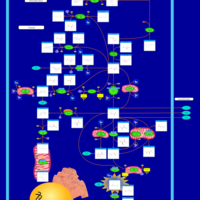 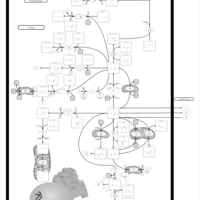 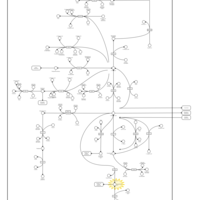 |
| Disulfiram Action Pathway | 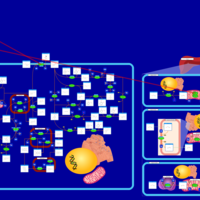 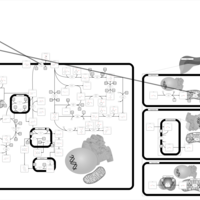 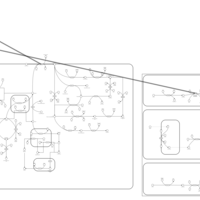 |
| Transfer of Acetyl Groups into Mitochondria | 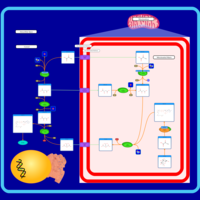 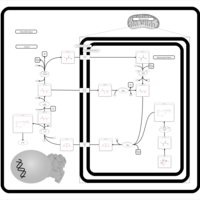 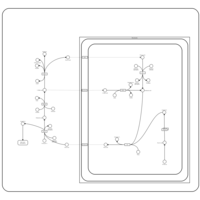 |
| Tyrosinemia, transient, of the newborn | 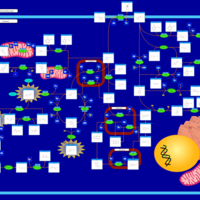 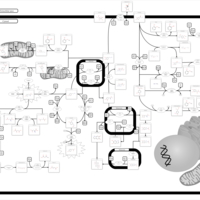 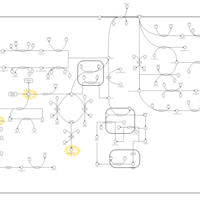 |
| Dopamine beta-hydroxylase deficiency |    |
| Creatine deficiency, guanidinoacetate methyltransferase deficiency |    |
| Hyperornithinemia with gyrate atrophy (HOGA) |    |
| Hyperornithinemia-hyperammonemia-homocitrullinuria [HHH-syndrome] |    |
| L-arginine:glycine amidinotransferase deficiency |    |
| Monoamine oxidase-a deficiency (MAO-A) |    |
| Congenital lactic acidosis | 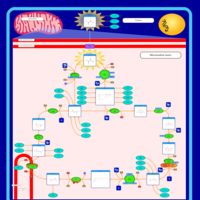 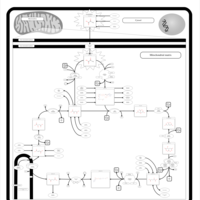 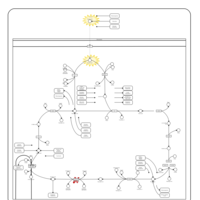 |
| Fumarase deficiency | 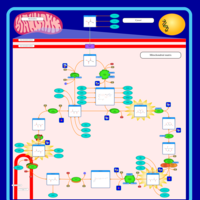 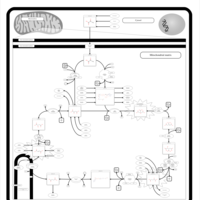 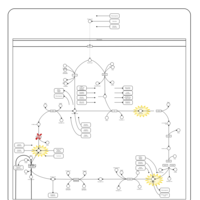 |
| Mitochondrial complex II deficiency |   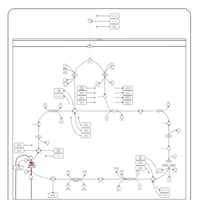 |
| 2-ketoglutarate dehydrogenase complex deficiency | 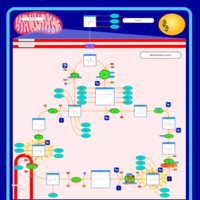 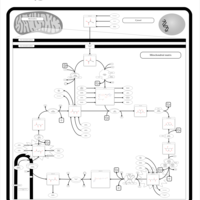 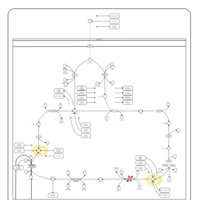 |
| Pyruvate dehydrogenase deficiency (E3) | 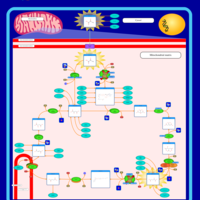 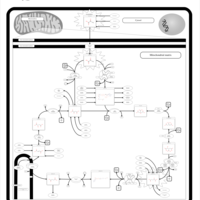 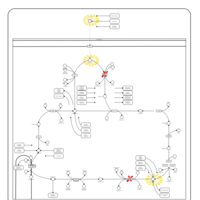 |
| Pyruvate dehydrogenase deficiency (E2) | 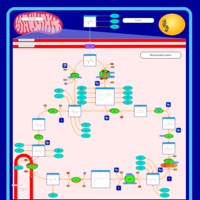 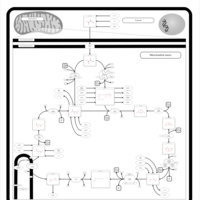 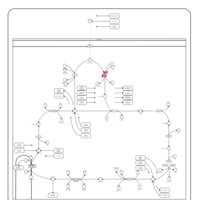 |
| Primary hyperoxaluria II, PH2 | 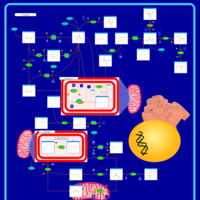 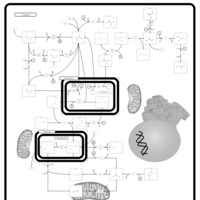  |
| Pyruvate kinase deficiency | 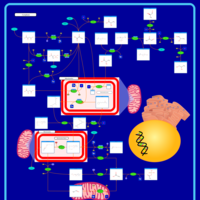 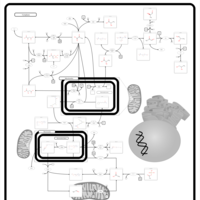 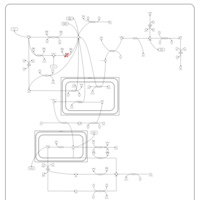 |
| Phosphoenolpyruvate carboxykinase deficiency 1 (PEPCK1) | 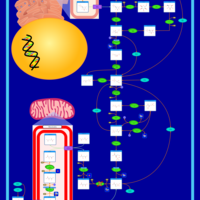 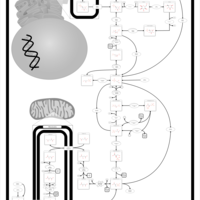 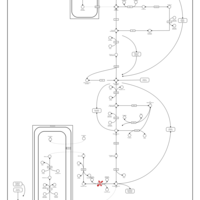 |
| Fructose-1,6-diphosphatase deficiency | 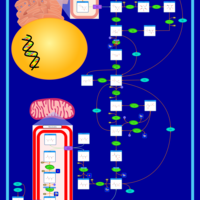 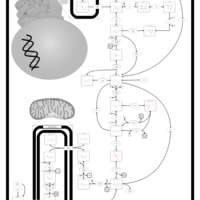 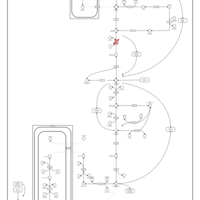 |
| Triosephosphate isomerase | 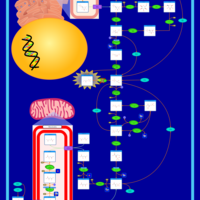 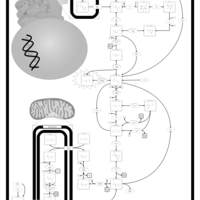 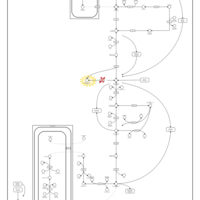 |
| Succinic semialdehyde dehydrogenase deficiency | 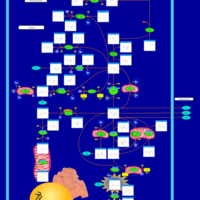 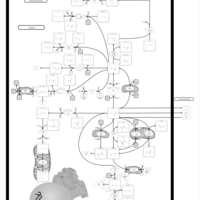 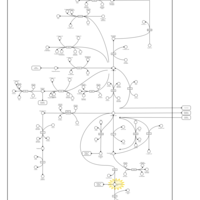 |
| Glycogenosis, Type IB |    |
| Glycogenosis, Type IC |    |
| Glycogenosis, Type IA. Von gierke disease |    |
| Warburg Effect | 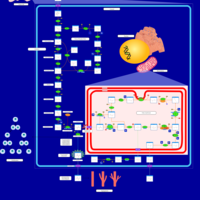 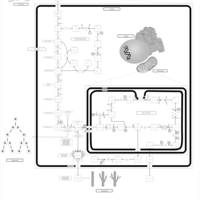 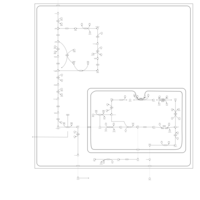 |
| The oncogenic action of 2-hydroxyglutarate | 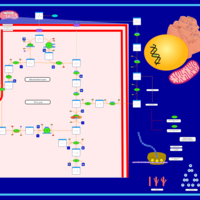 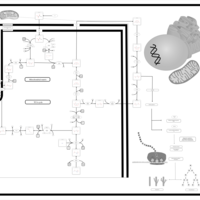 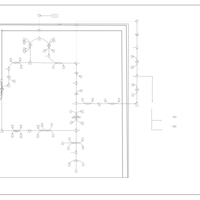 |
| The Oncogenic Action of Succinate | 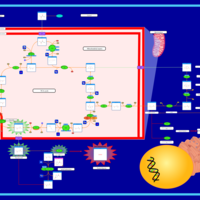  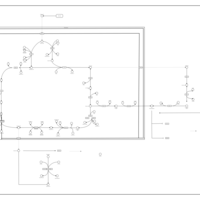 |
| The Oncogenic Action of Fumarate | 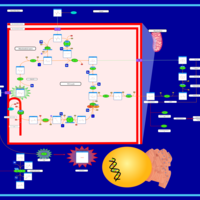 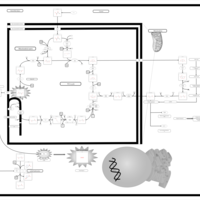  |
| Glutaminolysis and Cancer | 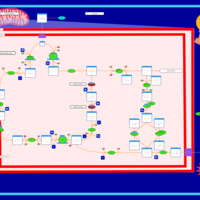 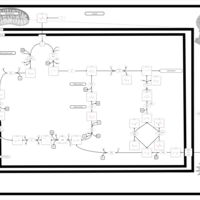 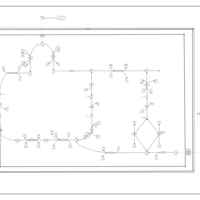 |
| The oncogenic action of L-2-hydroxyglutarate in Hydroxygluaricaciduria | 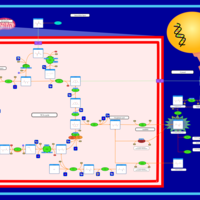 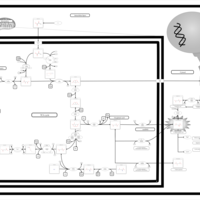 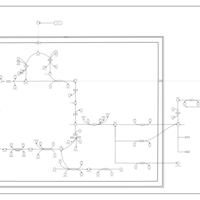 |
| The oncogenic action of D-2-hydroxyglutarate in Hydroxygluaricaciduria | 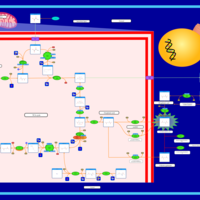 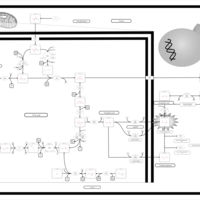 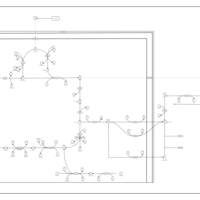 |
