| Oxidative phosphorylation | 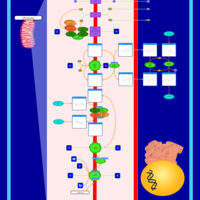   |
| Arginine and proline metabolism |  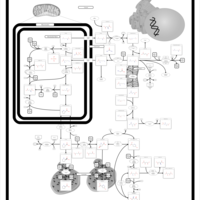 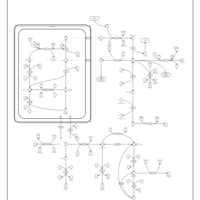 |
| Valine, leucine and isoleucine degradation |  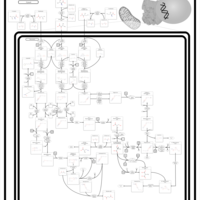 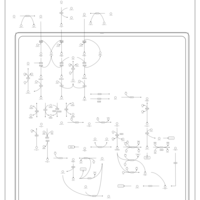 |
| Oxidation of Branched Chain Fatty Acids | 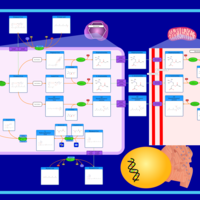 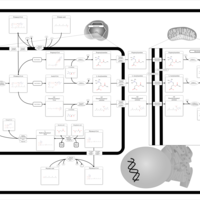  |
| Citric Acid Cycle |  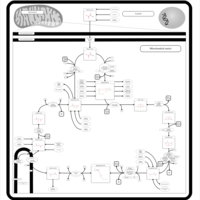 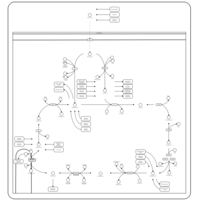 |
| Ketone Body Metabolism | 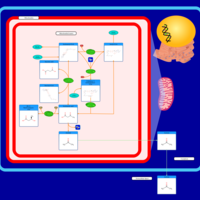  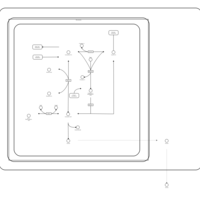 |
| Glutamate Metabolism | 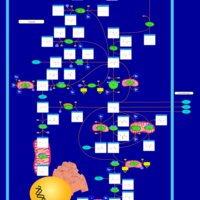 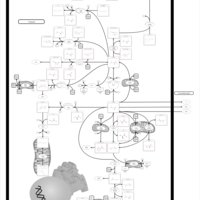 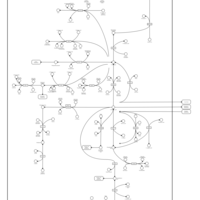 |
| Butyrate Metabolism | 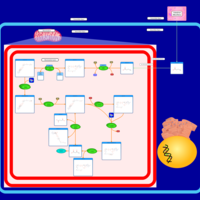 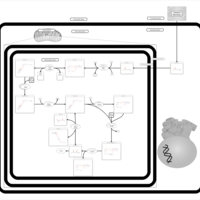 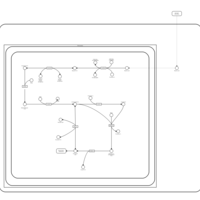 |
| 2-Hydroxyglutric Aciduria (D And L Form) | 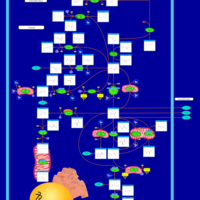 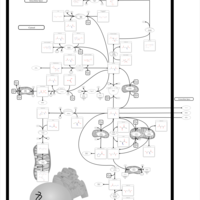 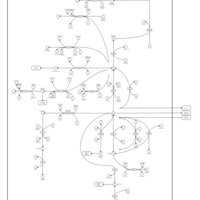 |
| 2-Methyl-3-Hydroxybutryl CoA Dehydrogenase Deficiency | 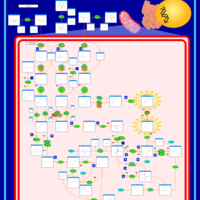 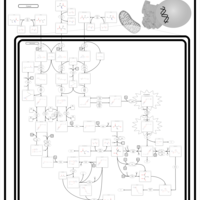 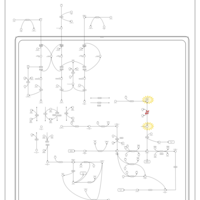 |
| 3-Hydroxy-3-Methylglutaryl-CoA Lyase Deficiency | 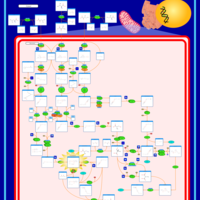 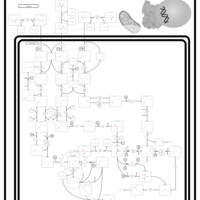 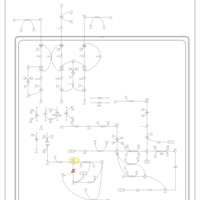 |
| 3-Methylglutaconic Aciduria Type I |  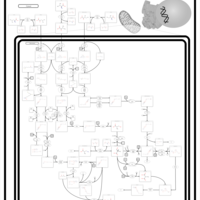 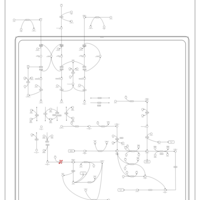 |
| 3-Methylglutaconic Aciduria Type III |  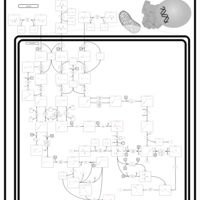 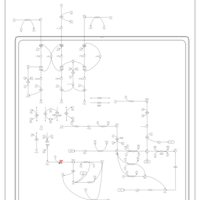 |
| 3-Methylglutaconic Aciduria Type IV | 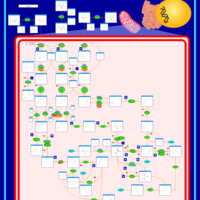 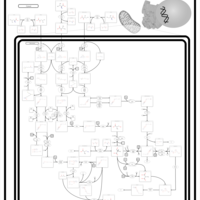  |
| Beta-Ketothiolase Deficiency | 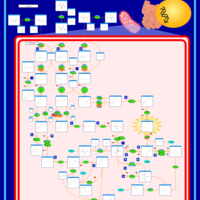 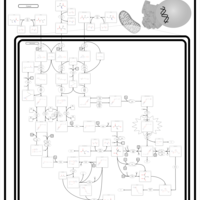 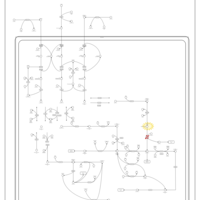 |
| Guanidinoacetate Methyltransferase Deficiency (GAMT Deficiency) |    |
| Maple Syrup Urine Disease |    |
| Methylmalonic Aciduria |    |
| Prolidase Deficiency (PD) | 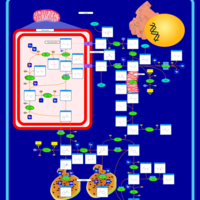 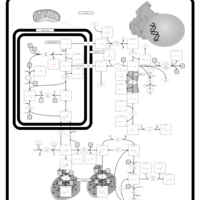 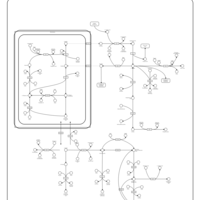 |
| Prolinemia Type II |   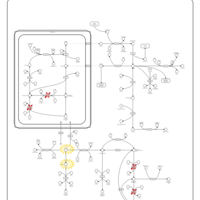 |
| Propionic Acidemia | 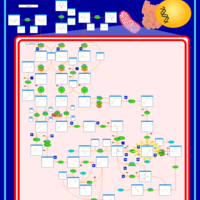 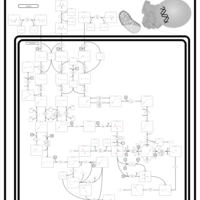 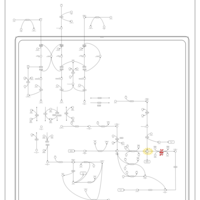 |
| 3-Methylcrotonyl Coa Carboxylase Deficiency Type I |  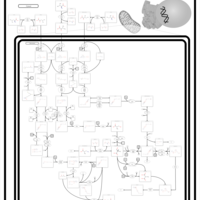 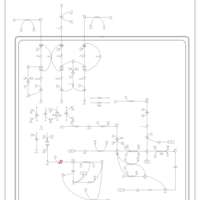 |
| Isovaleric Aciduria |    |
| 4-Hydroxybutyric Aciduria/Succinic Semialdehyde Dehydrogenase Deficiency | 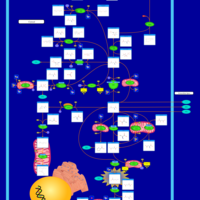 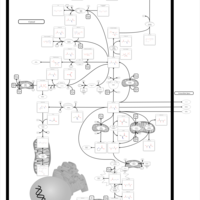 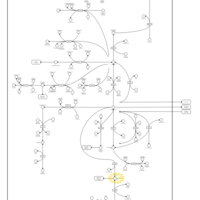 |
| Hyperinsulinism-Hyperammonemia Syndrome | 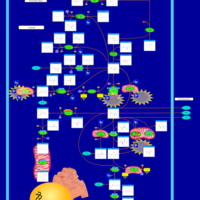 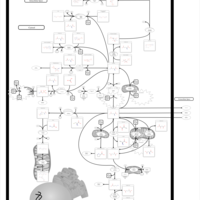 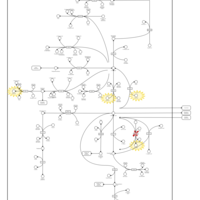 |
| Hyperprolinemia Type II | 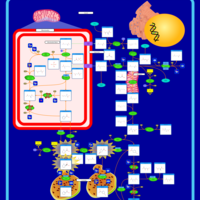 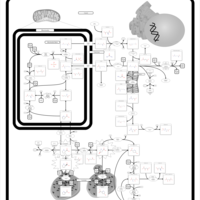 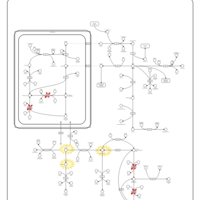 |
| Hyperprolinemia Type I | 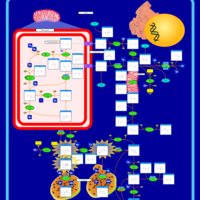 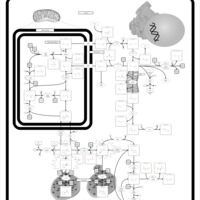 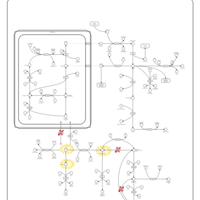 |
| Arginine: Glycine Amidinotransferase Deficiency (AGAT Deficiency) | 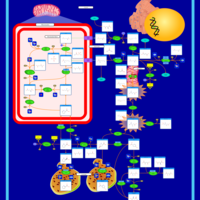 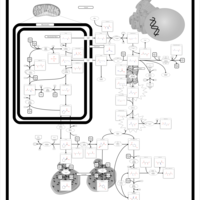 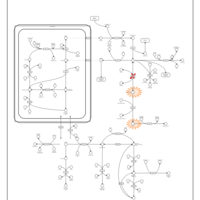 |
| Ornithine Aminotransferase Deficiency (OAT Deficiency) |    |
| Methylmalonate Semialdehyde Dehydrogenase Deficiency |    |
| Homocarnosinosis |  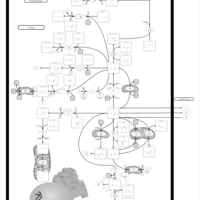 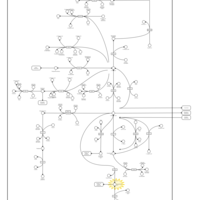 |
| Phytanic Acid Peroxisomal Oxidation | 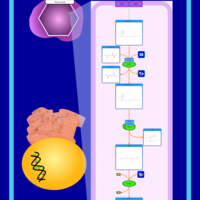 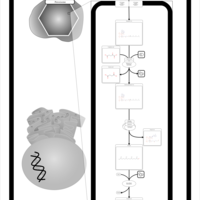 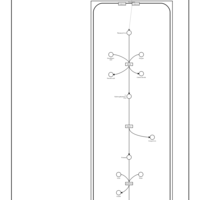 |
| Refsum Disease | 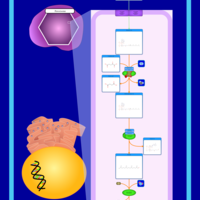 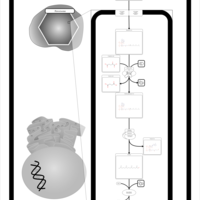  |
| Carnitine Synthesis | 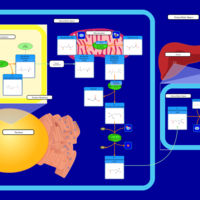 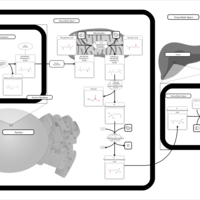 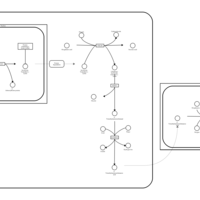 |
| Creatine deficiency, guanidinoacetate methyltransferase deficiency |    |
| Hyperornithinemia with gyrate atrophy (HOGA) |    |
| Hyperornithinemia-hyperammonemia-homocitrullinuria [HHH-syndrome] |    |
| L-arginine:glycine amidinotransferase deficiency |    |
| 3-hydroxyisobutyric acid dehydrogenase deficiency |   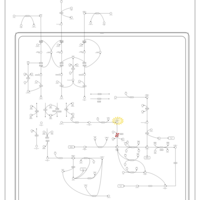 |
| 3-hydroxyisobutyric aciduria |  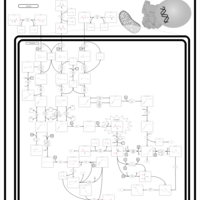 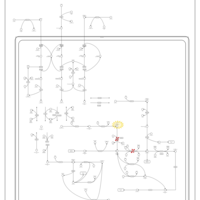 |
| Isobutyryl-coa dehydrogenase deficiency |    |
| Isovaleric acidemia |    |
| Congenital lactic acidosis | 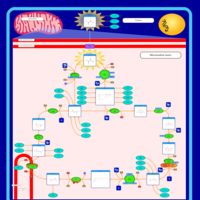 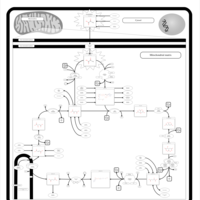 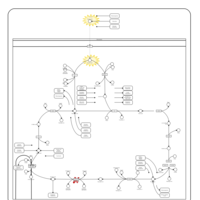 |
| Fumarase deficiency | 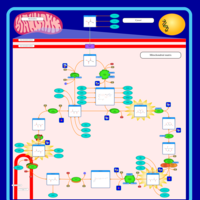 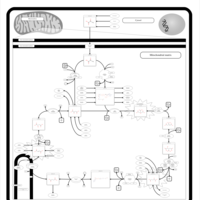 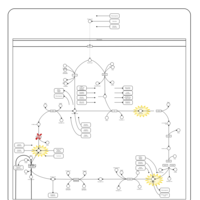 |
| Mitochondrial complex II deficiency | 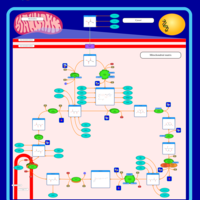 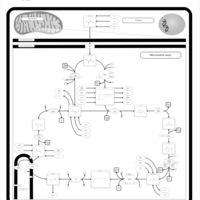 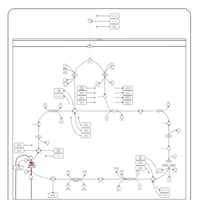 |
| 2-ketoglutarate dehydrogenase complex deficiency |  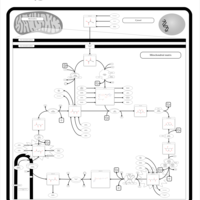 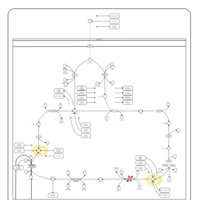 |
| Pyruvate dehydrogenase deficiency (E3) | 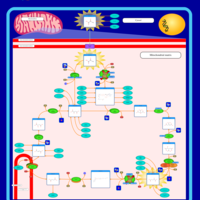 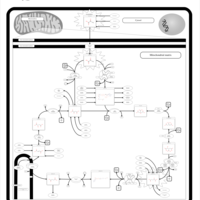 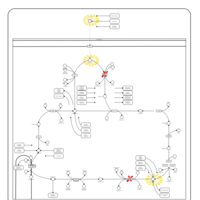 |
| Pyruvate dehydrogenase deficiency (E2) | 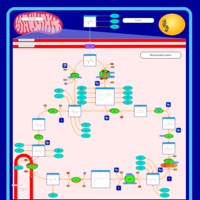 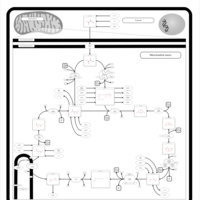 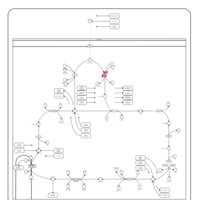 |
| Succinic semialdehyde dehydrogenase deficiency | 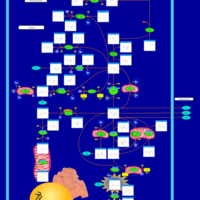 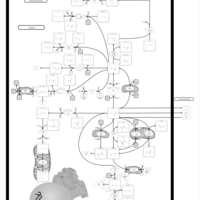 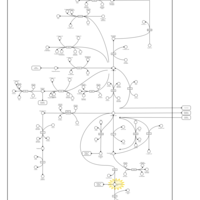 |
| Succinyl CoA: 3-ketoacid CoA transferase deficiency |    |
| Warburg Effect | 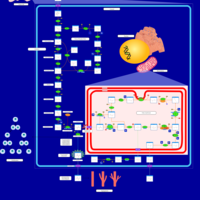 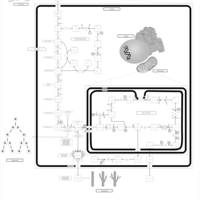  |
| The oncogenic action of 2-hydroxyglutarate | 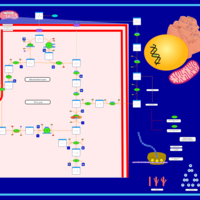 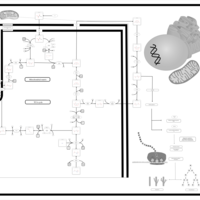 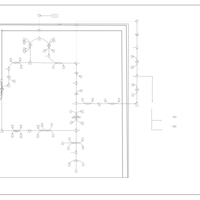 |
| The Oncogenic Action of Succinate | 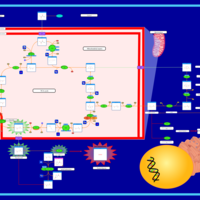  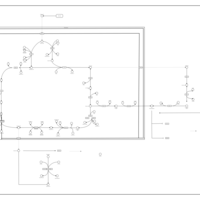 |
| The Oncogenic Action of Fumarate | 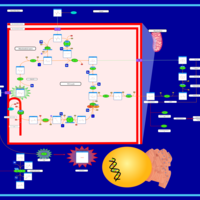 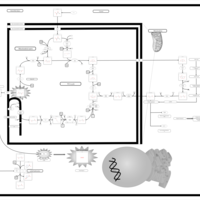  |
| Glutaminolysis and Cancer | 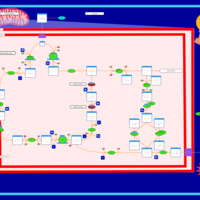 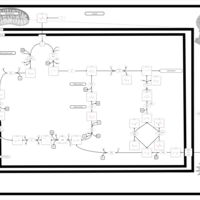 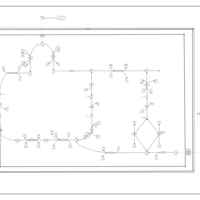 |
| The oncogenic action of L-2-hydroxyglutarate in Hydroxygluaricaciduria | 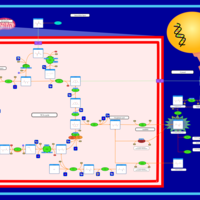 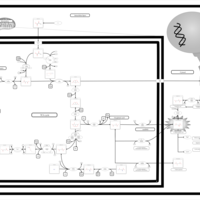 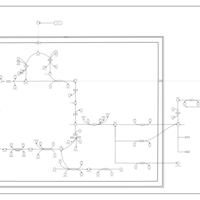 |
| The oncogenic action of D-2-hydroxyglutarate in Hydroxygluaricaciduria | 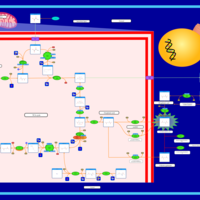 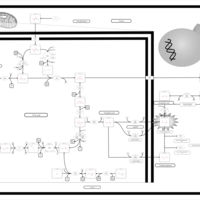 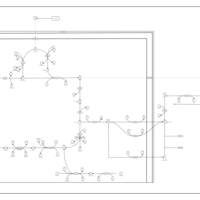 |
