| Nitrogen metabolism |  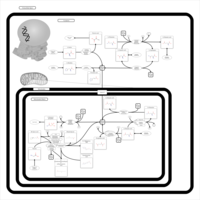 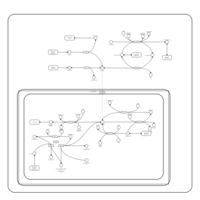 |
| Primary bile acid biosynthesis | 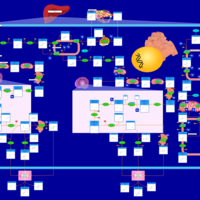 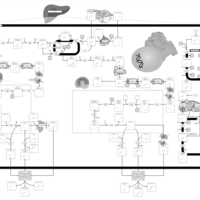 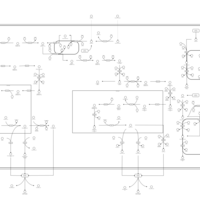 |
| Glutathione metabolism | 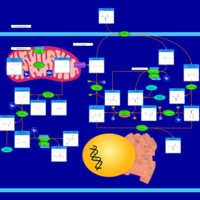 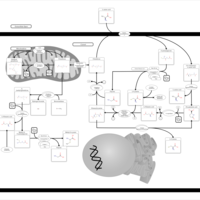 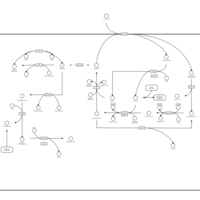 |
| Glycine, serine and threonine metabolism | 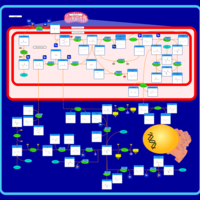 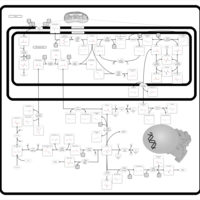 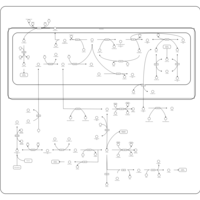 |
| Alanine, aspartate and glutamate metabolism | 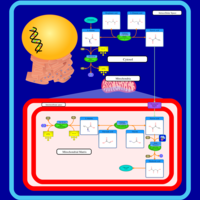 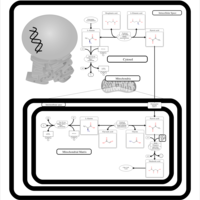  |
| Arginine and proline metabolism | 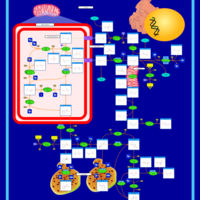 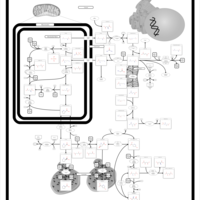 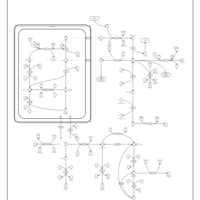 |
| Purine metabolism |  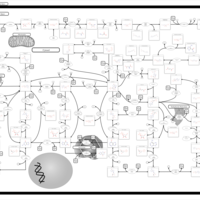 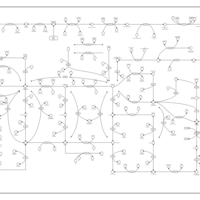 |
| Porphyrin Metabolism | 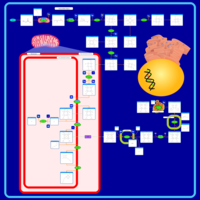 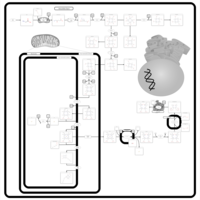 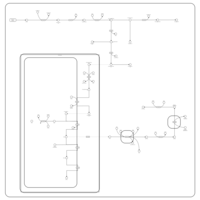 |
| Methionine Metabolism |  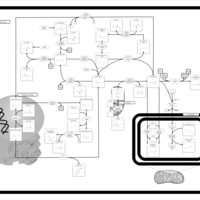 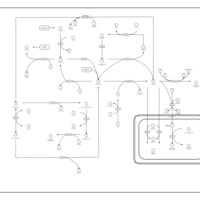 |
| Glutamate Metabolism | 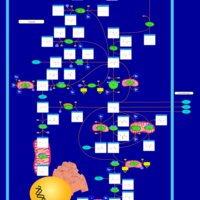 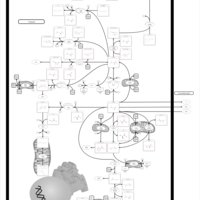 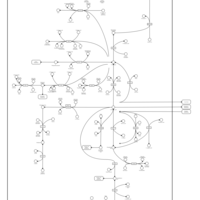 |
| 2-Hydroxyglutric Aciduria (D And L Form) | 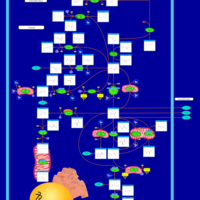 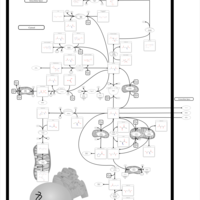 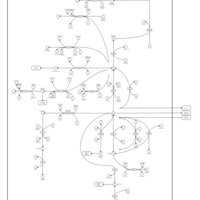 |
| 5-Oxoprolinuria | 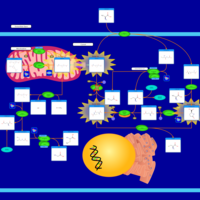 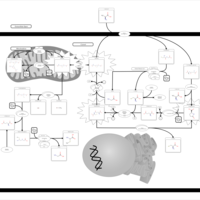 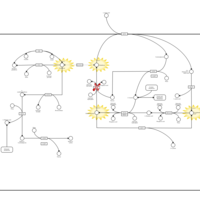 |
| Adenosine Deaminase Deficiency |  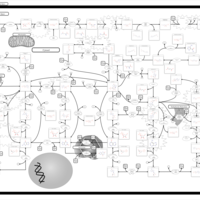  |
| Adenylosuccinate Lyase Deficiency |  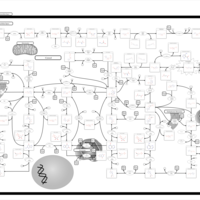 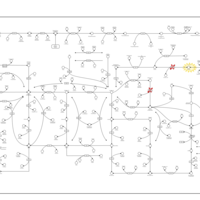 |
| AICA-Ribosiduria | 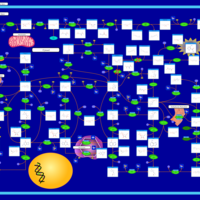 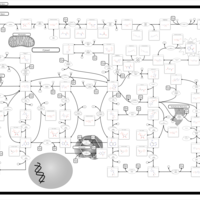 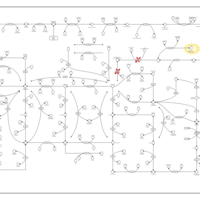 |
| Cystathionine Beta-Synthase Deficiency | 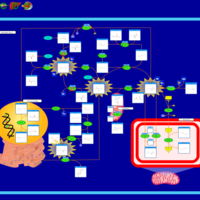 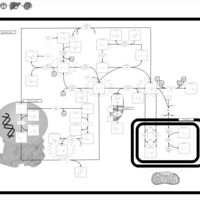 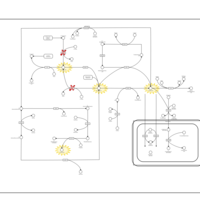 |
| Dihydropyrimidine Dehydrogenase Deficiency (DHPD) | 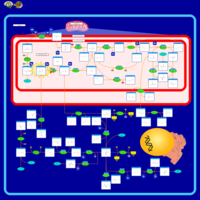 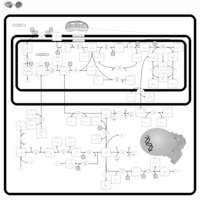 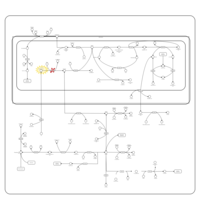 |
| Gamma-Glutamyltransferase Deficiency | 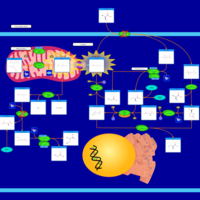 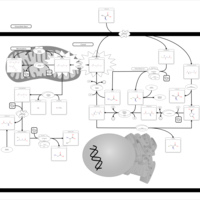 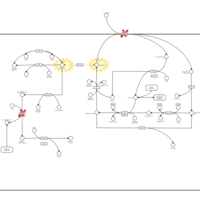 |
| Guanidinoacetate Methyltransferase Deficiency (GAMT Deficiency) |    |
| Molybdenum Cofactor Deficiency |    |
| Prolidase Deficiency (PD) | 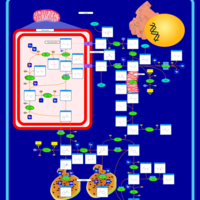 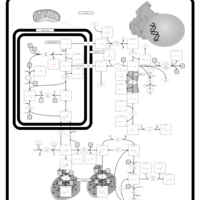 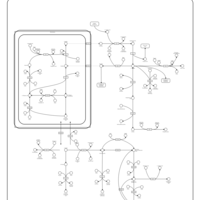 |
| Prolinemia Type II |   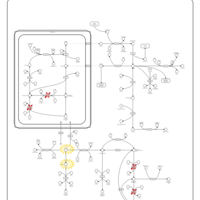 |
| Purine Nucleoside Phosphorylase Deficiency |    |
| S-Adenosylhomocysteine (SAH) Hydrolase Deficiency | 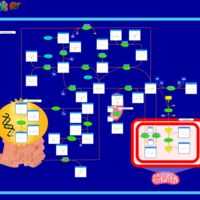 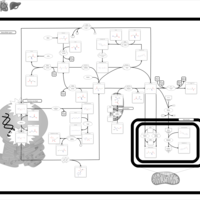 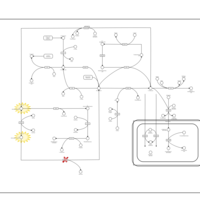 |
| Xanthine Dehydrogenase Deficiency (Xanthinuria) | 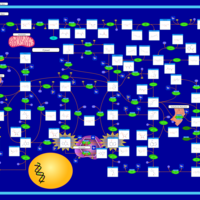 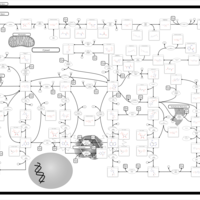  |
| Methionine Adenosyltransferase Deficiency |    |
| Glycine N-methyltransferase Deficiency | 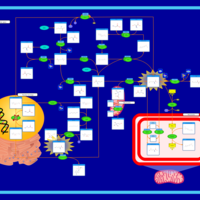 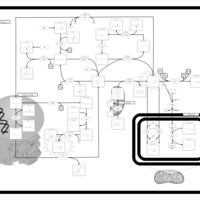 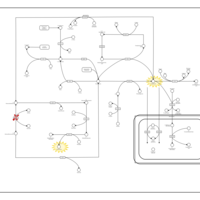 |
| Non Ketotic Hyperglycinemia | 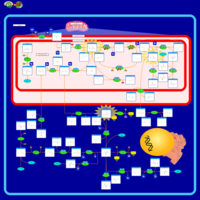 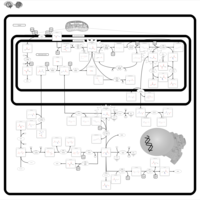 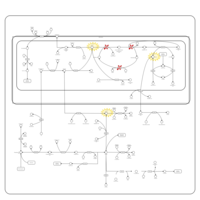 |
| Dimethylglycine Dehydrogenase Deficiency | 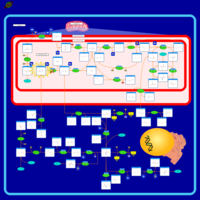 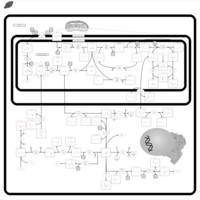  |
| 4-Hydroxybutyric Aciduria/Succinic Semialdehyde Dehydrogenase Deficiency | 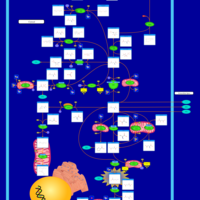 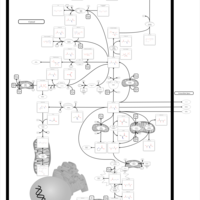 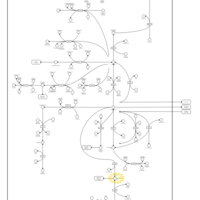 |
| Sarcosinemia | 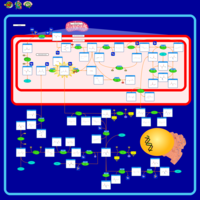 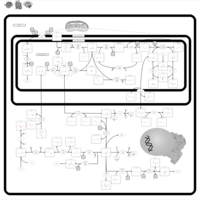 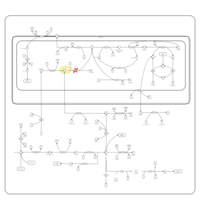 |
| Lactic Acidemia | 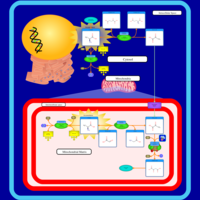 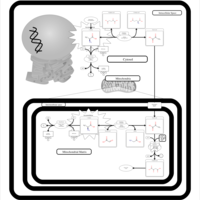 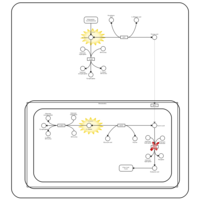 |
| Congenital Bile Acid Synthesis Defect Type II | 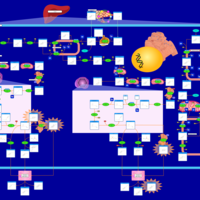 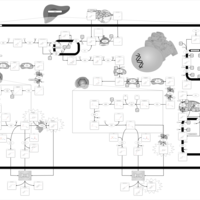 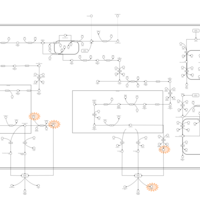 |
| Cerebrotendinous Xanthomatosis (CTX) |    |
| Zellweger Syndrome | 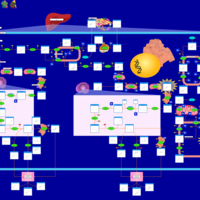 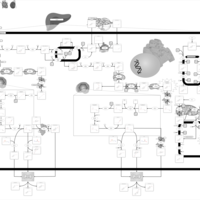 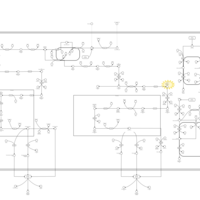 |
| Familial Hypercholanemia (FHCA) |  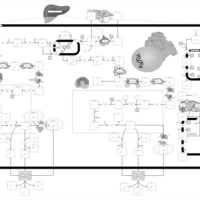 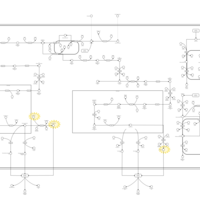 |
| Congenital Bile Acid Synthesis Defect Type III | 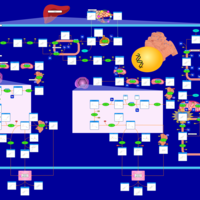 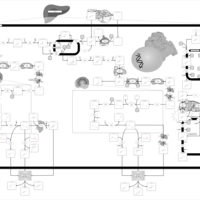 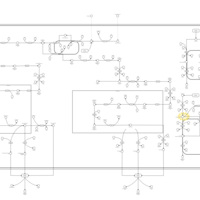 |
| Glutathione Synthetase Deficiency | 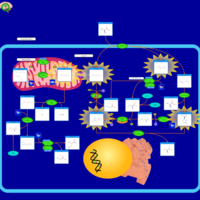 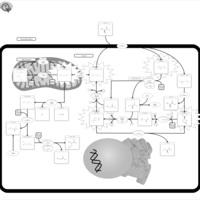 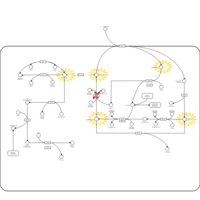 |
| Hyperinsulinism-Hyperammonemia Syndrome | 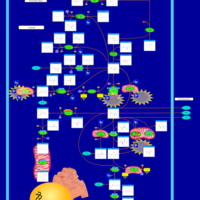 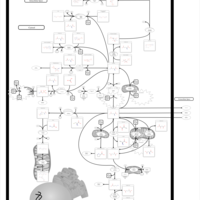 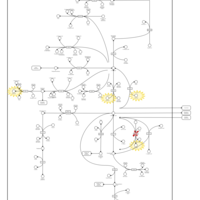 |
| Methylenetetrahydrofolate Reductase Deficiency (MTHFRD) |    |
| Hypermethioninemia |   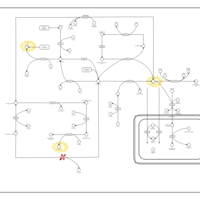 |
| Hereditary Coproporphyria (HCP) | 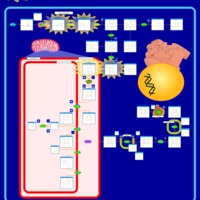 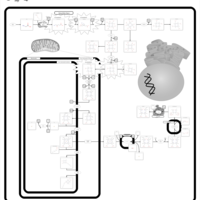 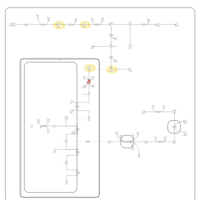 |
| Acute Intermittent Porphyria | 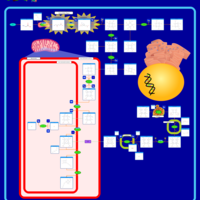   |
| Congenital Erythropoietic Porphyria (CEP) or Gunther Disease | 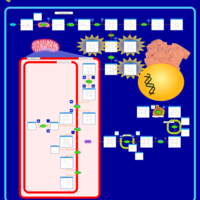 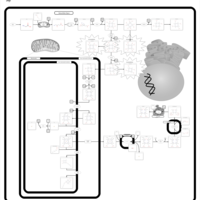 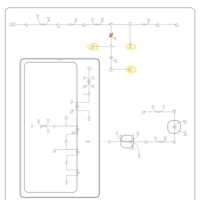 |
| Porphyria Variegata (PV) | 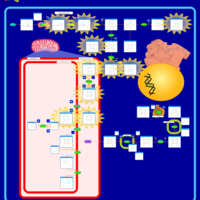 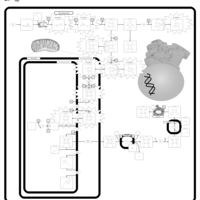 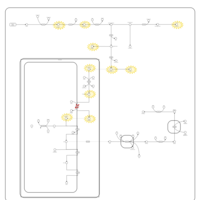 |
| Pyruvate Carboxylase Deficiency | 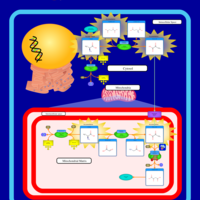 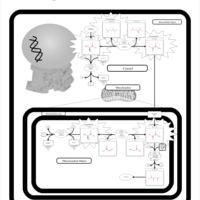 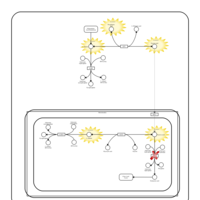 |
| Primary Hyperoxaluria Type I | 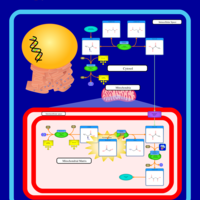 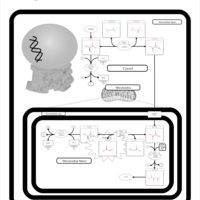 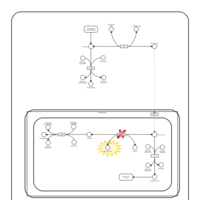 |
| Hyperprolinemia Type II | 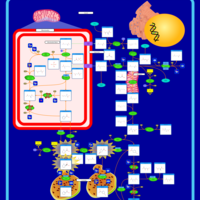 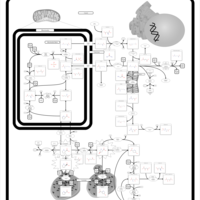 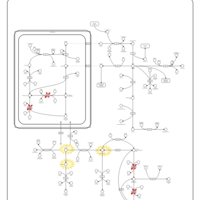 |
| Hyperprolinemia Type I | 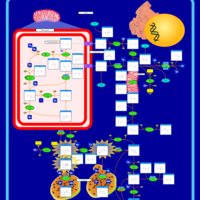 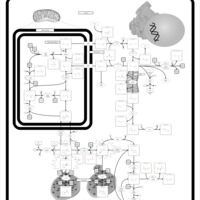 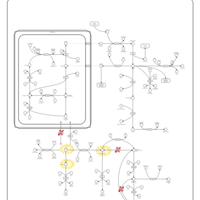 |
| Arginine: Glycine Amidinotransferase Deficiency (AGAT Deficiency) | 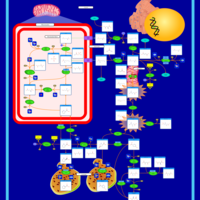 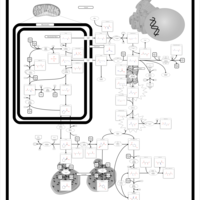 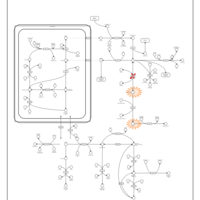 |
| Ornithine Aminotransferase Deficiency (OAT Deficiency) |    |
| Lesch-Nyhan Syndrome (LNS) | 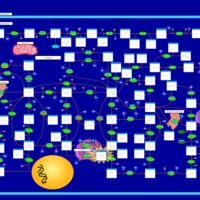 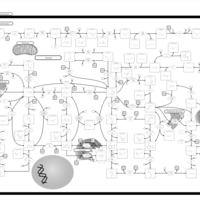 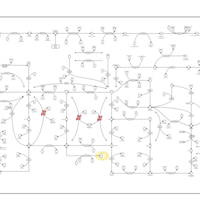 |
| Gout or Kelley-Seegmiller Syndrome | 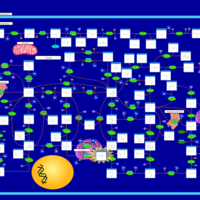 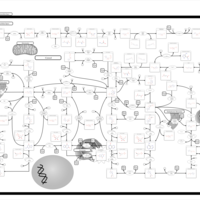 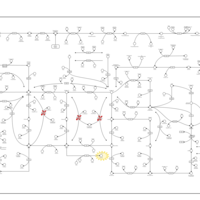 |
| Homocarnosinosis | 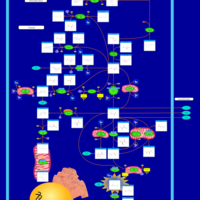 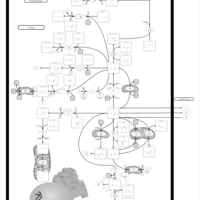 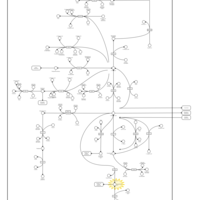 |
| Azathioprine Action Pathway | 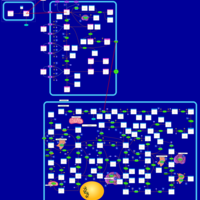   |
| Mercaptopurine Action Pathway |  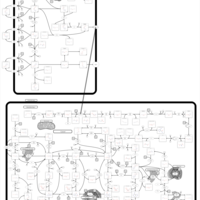 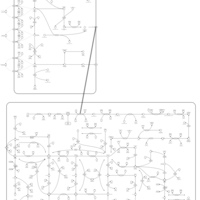 |
| Thioguanine Action Pathway | 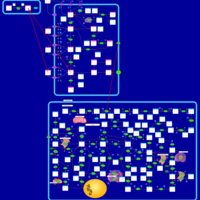  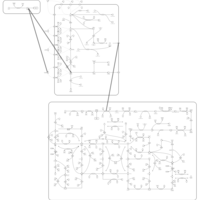 |
| Carnitine Synthesis | 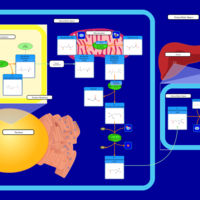 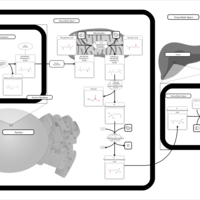 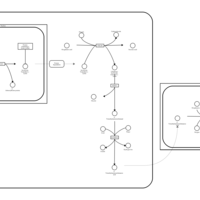 |
| Dimethylglycine Dehydrogenase Deficiency |    |
| Hyperglycinemia, non-ketotic |    |
| 5-oxoprolinase deficiency | 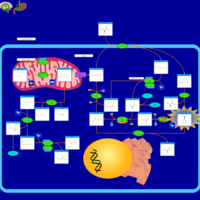 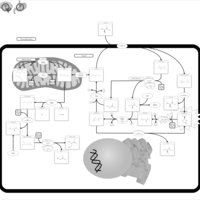 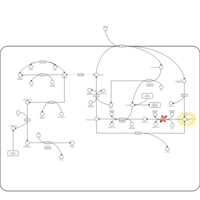 |
| Gamma-glutamyl-transpeptidase deficiency | 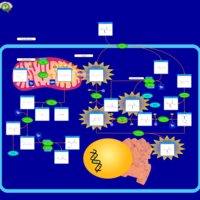 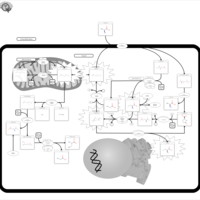 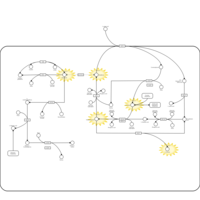 |
| Creatine deficiency, guanidinoacetate methyltransferase deficiency |    |
| Hyperornithinemia with gyrate atrophy (HOGA) |    |
| Hyperornithinemia-hyperammonemia-homocitrullinuria [HHH-syndrome] |    |
| L-arginine:glycine amidinotransferase deficiency |    |
| Xanthinuria type I |  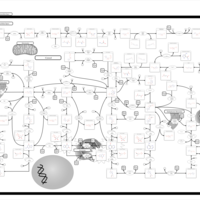 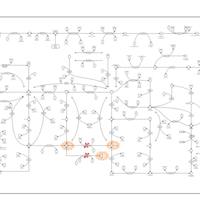 |
| Xanthinuria type II |  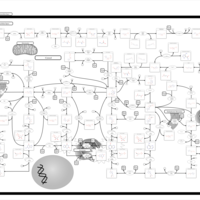  |
| Adenine phosphoribosyltransferase deficiency (APRT) | 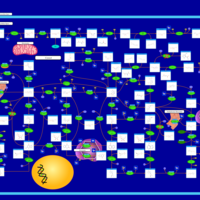 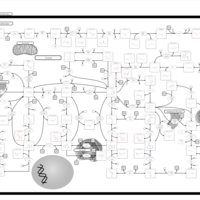  |
| Mitochondrial DNA depletion syndrome |    |
| Myoadenylate deaminase deficiency |    |
| Succinic semialdehyde dehydrogenase deficiency | 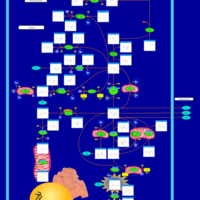 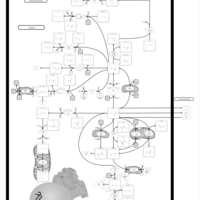 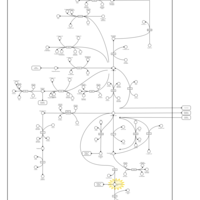 |
| Homocystinuria-megaloblastic anemia due to defect in cobalamin metabolism, cblG complementation type |    |
| 27-Hydroxylase Deficiency | 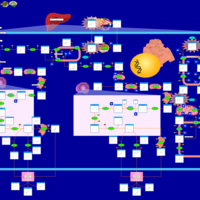 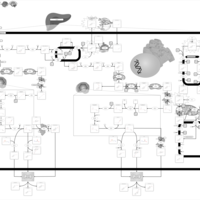 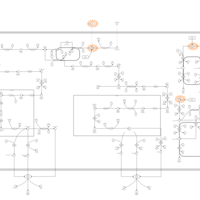 |
| 3-Phosphoglycerate dehydrogenase deficiency | 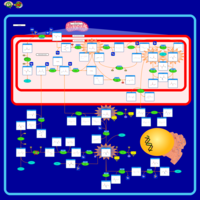 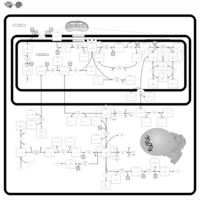 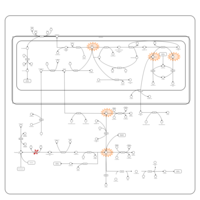 |
| Sarcosine Oncometabolite Pathway |  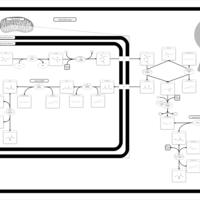  |
