| Glutathione metabolism | 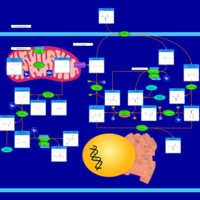 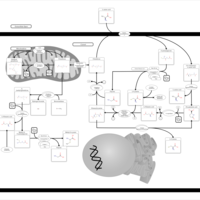 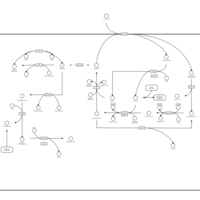 |
| Arginine and proline metabolism | 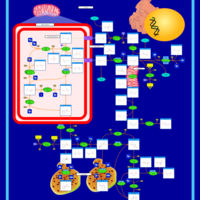 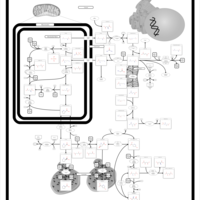 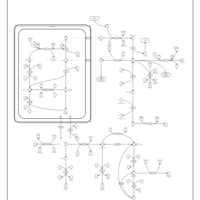 |
| beta-Alanine metabolism |   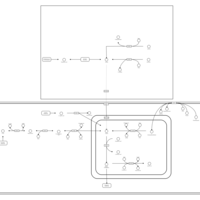 |
| One carbon pool by folate |   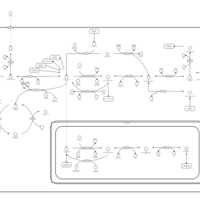 |
| Lysine degradation | 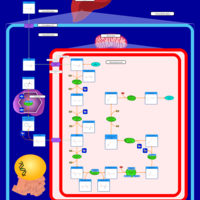 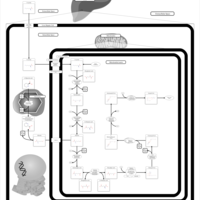 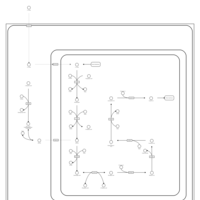 |
| Valine, leucine and isoleucine degradation |  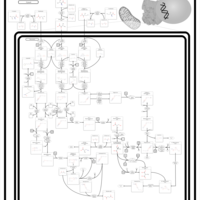 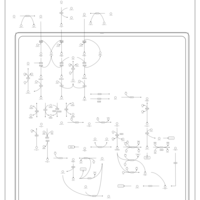 |
| Pyruvate metabolism |  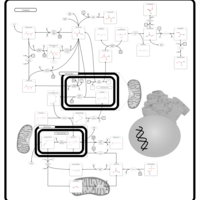  |
| Purine metabolism |  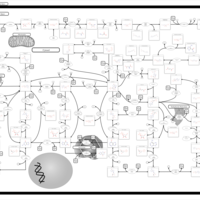 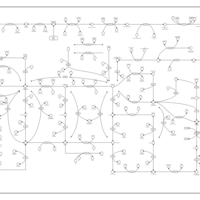 |
| Tyrosine metabolism | 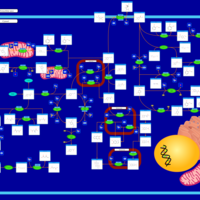 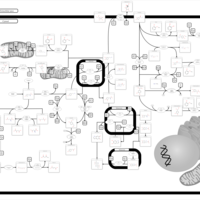 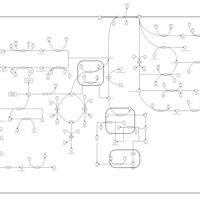 |
| Glycine, serine and threonine metabolism | 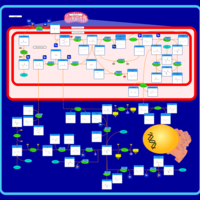 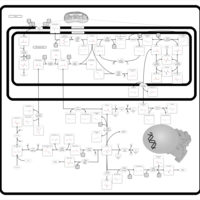 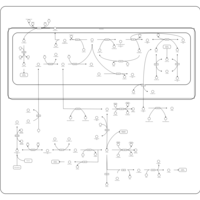 |
| Pyrimidine metabolism |  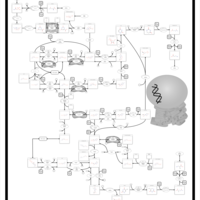  |
| Primary bile acid biosynthesis |  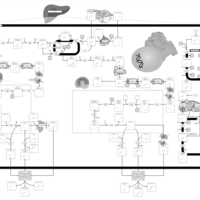 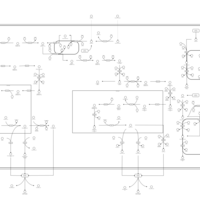 |
| Tryptophan metabolism |  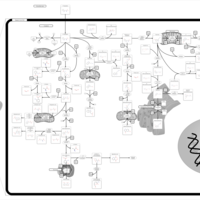 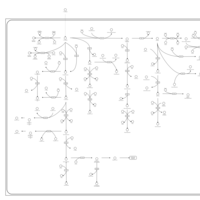 |
| Oxidative phosphorylation | 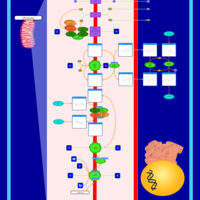 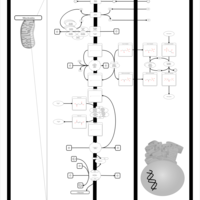 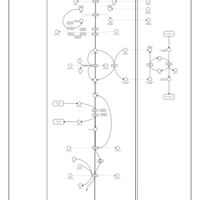 |
| Nicotinate and nicotinamide metabolism | 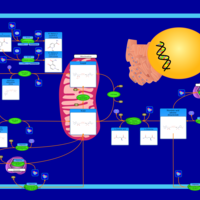 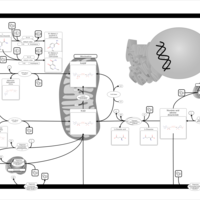 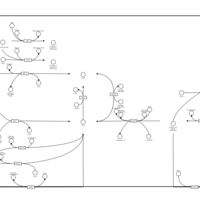 |
| Propanoate metabolism | 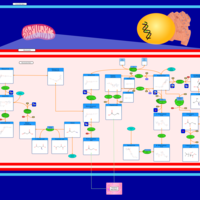 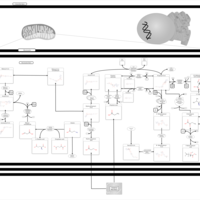 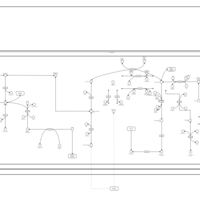 |
| Glycine, serine and threonine metabolism |  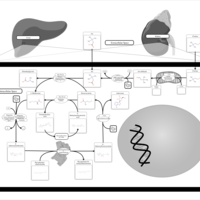  |
| Nitrogen metabolism |  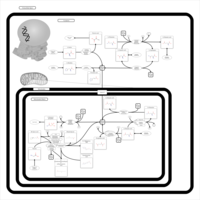 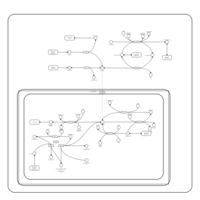 |
| Phenylalanine and Tyrosine Metabolism | 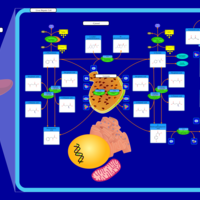 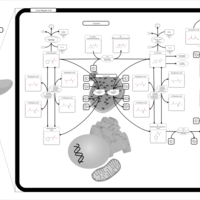 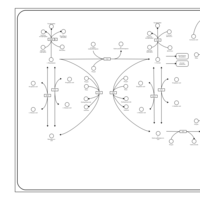 |
| Vitamin B6 Metabolism | 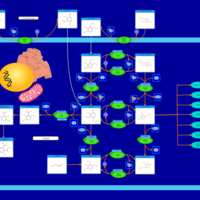 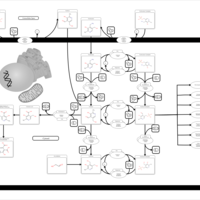 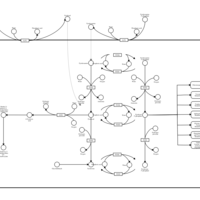 |
| Steroid Biosynthesis | 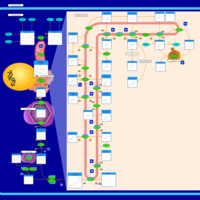 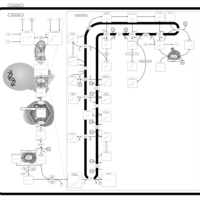 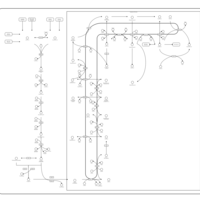 |
| Porphyrin Metabolism | 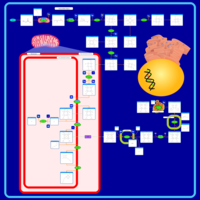 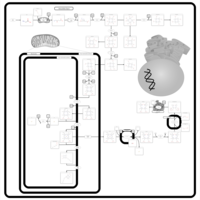 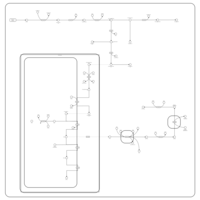 |
| Phospholipid Biosynthesis |  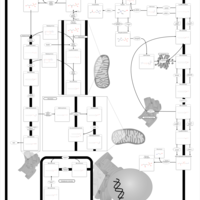 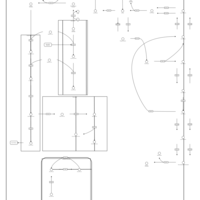 |
| Caffeine Metabolism | 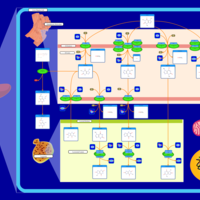 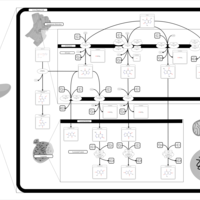 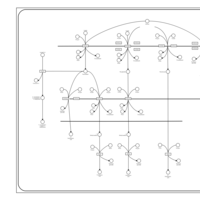 |
| Methionine Metabolism |   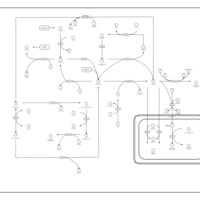 |
| D-Arginine and D-Ornithine Metabolism |    |
| Glycerolipid Metabolism |    |
| Histidine metabolism |    |
| Fatty acid Metabolism |    |
| Citric Acid Cycle |  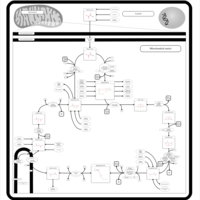 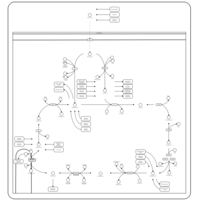 |
| Aspartate Metabolism | 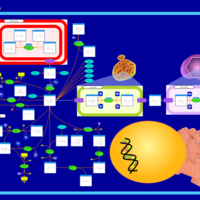 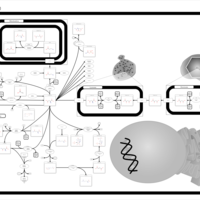  |
| Riboflavin Metabolism |    |
| Glutamate Metabolism |    |
| Butyrate Metabolism |  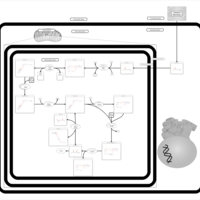 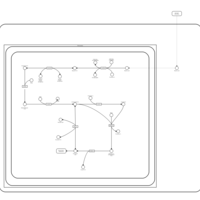 |
| Ibandronate Action Pathway | 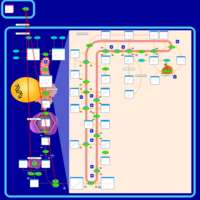 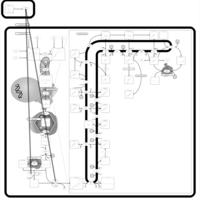 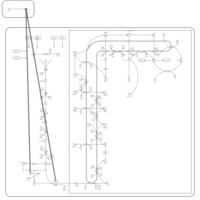 |
| Simvastatin Action Pathway | 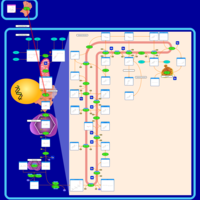 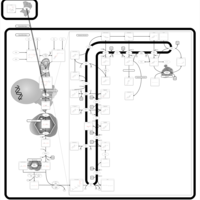 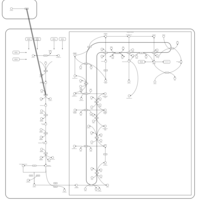 |
| Pravastatin Action Pathway | 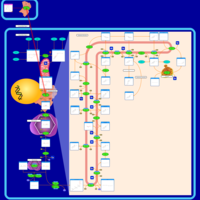 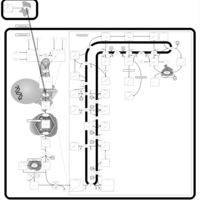  |
| Rosuvastatin Action Pathway | 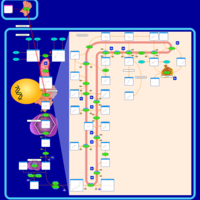 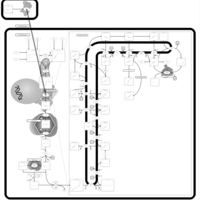 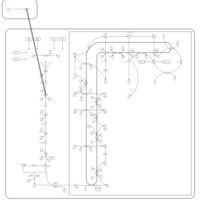 |
| Alendronate Action Pathway | 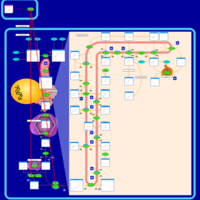 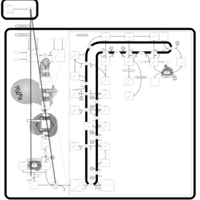 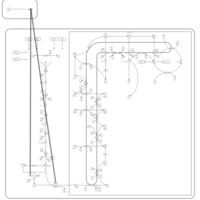 |
| Lovastatin Action Pathway | 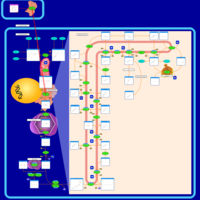 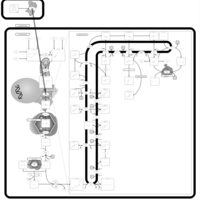 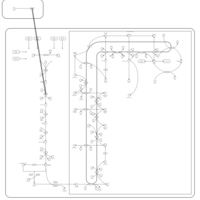 |
| Zoledronate Action Pathway | 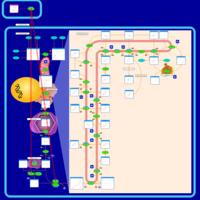 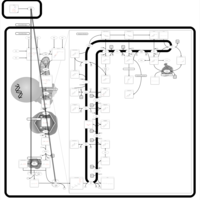 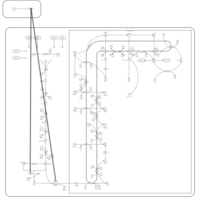 |
| Cerivastatin Action Pathway |    |
| Risedronate Action Pathway | 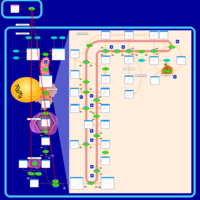 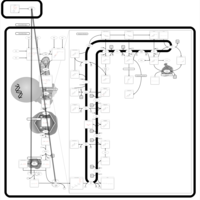 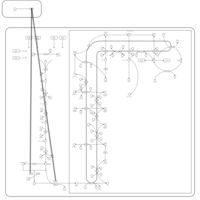 |
| Pamidronate Action Pathway | 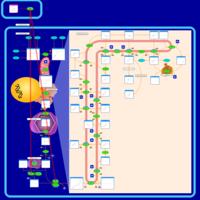 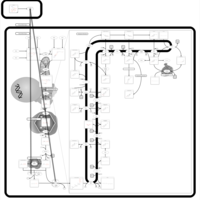 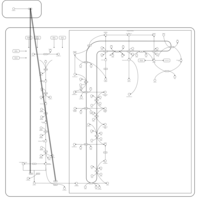 |
| Fluvastatin Action Pathway | 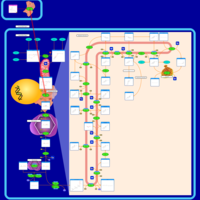 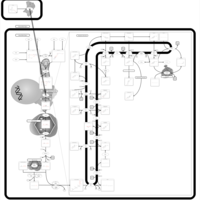 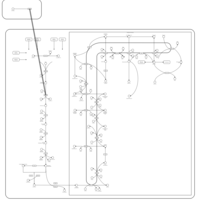 |
| Glycerol Phosphate Shuttle | 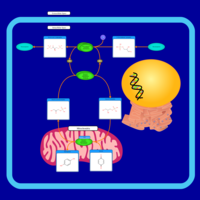   |
| Atorvastatin Action Pathway |    |
| 2-Hydroxyglutric Aciduria (D And L Form) | 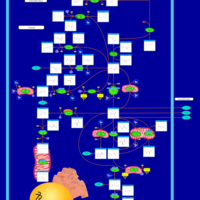 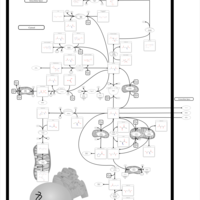 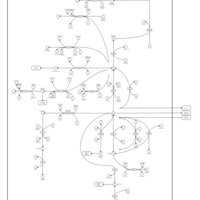 |
| 2-Methyl-3-Hydroxybutryl CoA Dehydrogenase Deficiency | 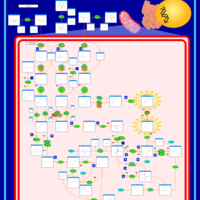   |
| 3-Hydroxy-3-Methylglutaryl-CoA Lyase Deficiency |   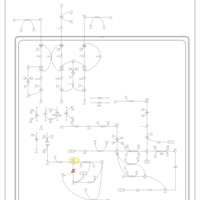 |
| 3-Methylglutaconic Aciduria Type I |   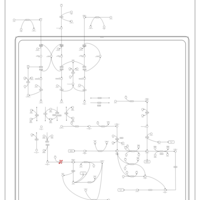 |
| 3-Methylglutaconic Aciduria Type III |  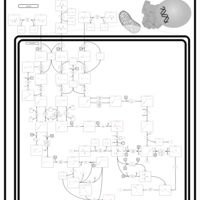 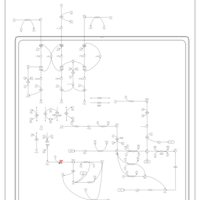 |
| 3-Methylglutaconic Aciduria Type IV | 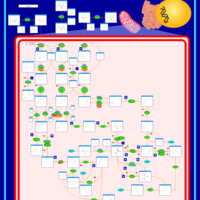 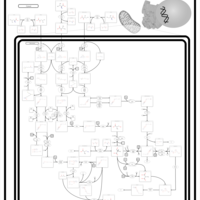 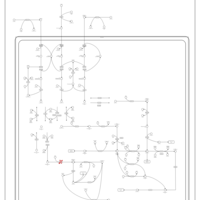 |
| 5-Oxoprolinuria | 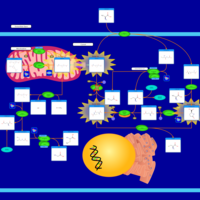 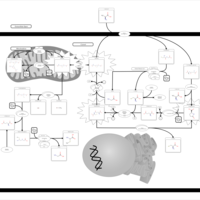 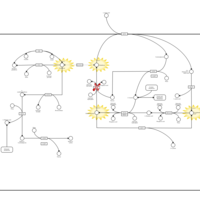 |
| Adenosine Deaminase Deficiency |  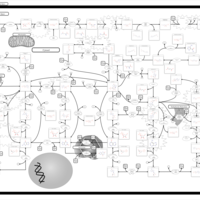 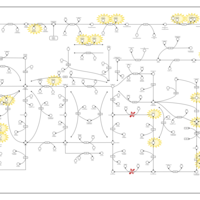 |
| Adenylosuccinate Lyase Deficiency |    |
| AICA-Ribosiduria |    |
| Alkaptonuria | 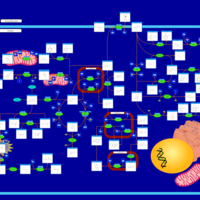 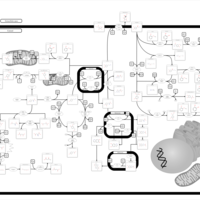 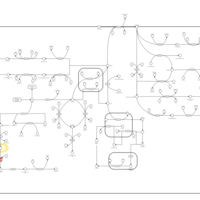 |
| Beta Ureidopropionase Deficiency |  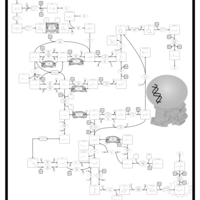 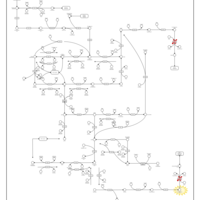 |
| Beta-Ketothiolase Deficiency | 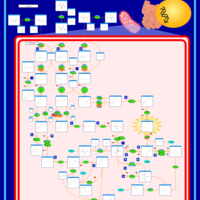   |
| Canavan Disease | 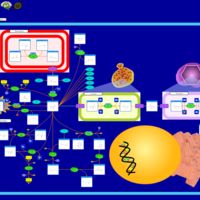 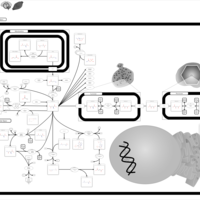 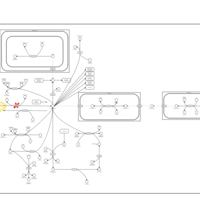 |
| Cystathionine Beta-Synthase Deficiency | 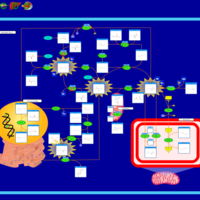 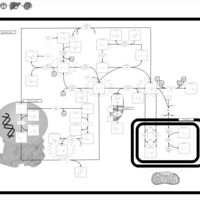 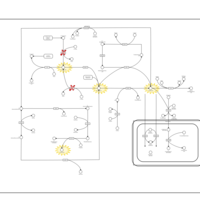 |
| Dihydropyrimidinase Deficiency |  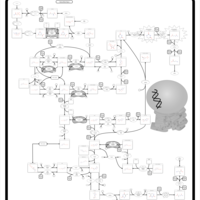 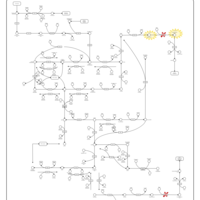 |
| Dihydropyrimidine Dehydrogenase Deficiency (DHPD) | 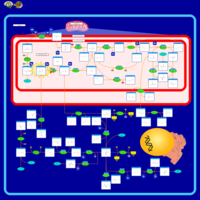 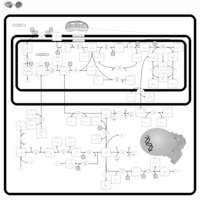 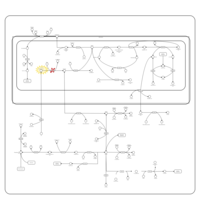 |
| Ethylmalonic Encephalopathy | 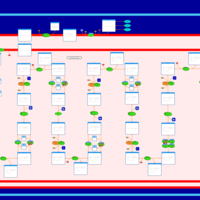  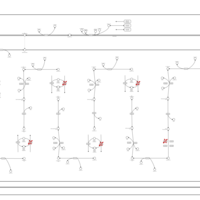 |
| Gamma-Glutamyltransferase Deficiency | 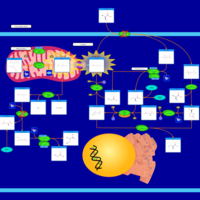 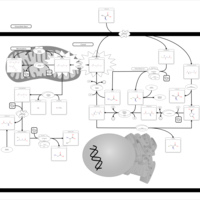 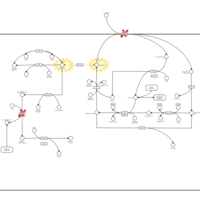 |
| Glutaric Aciduria Type I | 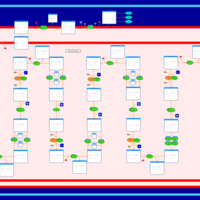  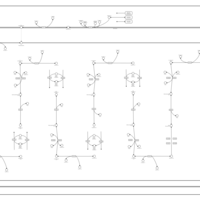 |
| Glutaric Aciduria Type I | 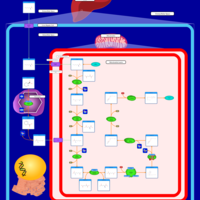 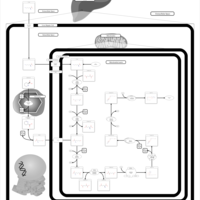 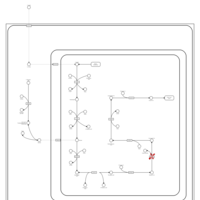 |
| Glycerol Kinase Deficiency | 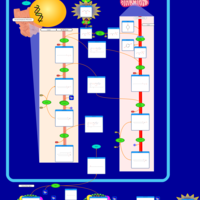 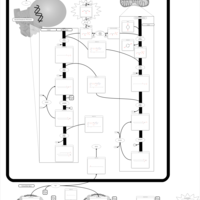 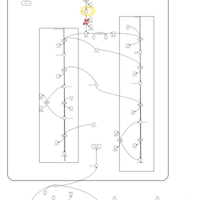 |
| Guanidinoacetate Methyltransferase Deficiency (GAMT Deficiency) |    |
| Hawkinsinuria | 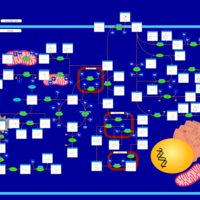  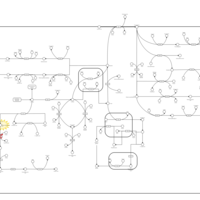 |
| Histidinemia | 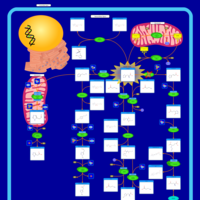 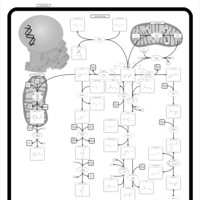  |
| Hypoacetylaspartia |    |
| Leigh Syndrome |  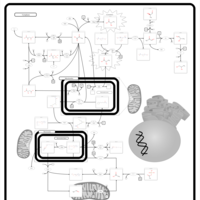 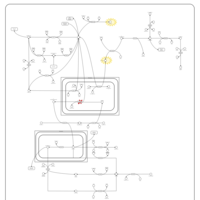 |
| Malonic Aciduria |    |
| Maple Syrup Urine Disease |    |
| Methylmalonic Aciduria |    |
| Methylmalonic Aciduria Due to Cobalamin-Related Disorders | 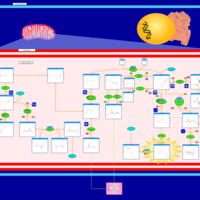 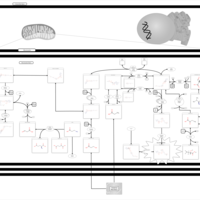 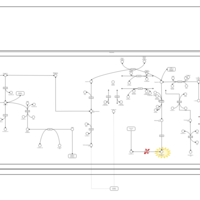 |
| MNGIE (Mitochondrial Neurogastrointestinal Encephalopathy) |  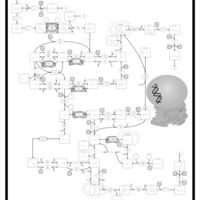 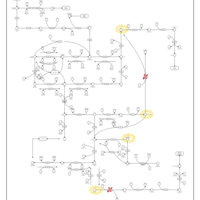 |
| Molybdenum Cofactor Deficiency |    |
| Phenylketonuria |   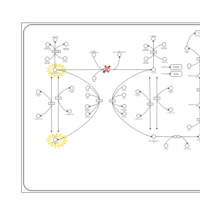 |
| Prolidase Deficiency (PD) | 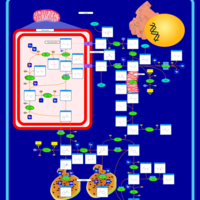 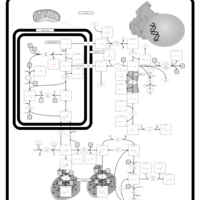  |
| Prolinemia Type II |    |
| Hypercholesterolemia |    |
| Purine Nucleoside Phosphorylase Deficiency |    |
| Pyruvate Dehydrogenase Complex Deficiency | 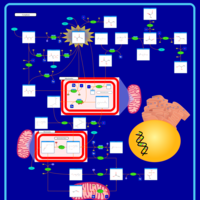 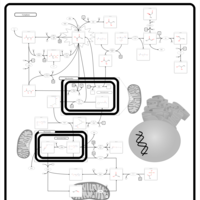 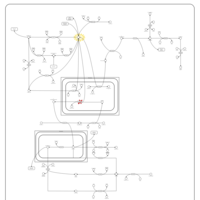 |
| S-Adenosylhomocysteine (SAH) Hydrolase Deficiency | 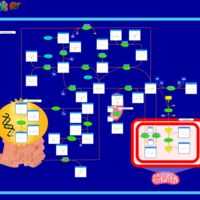 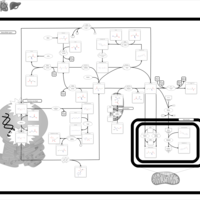 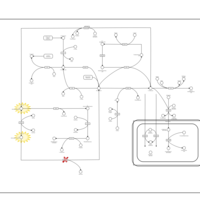 |
| Tyrosinemia Type I | 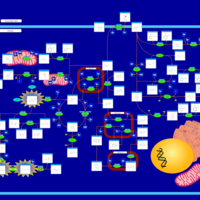 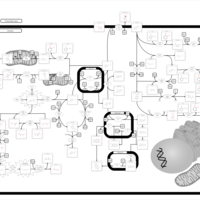 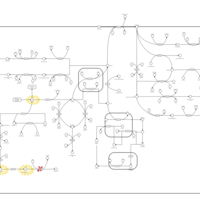 |
| UMP Synthase Deficiency (Orotic Aciduria) |  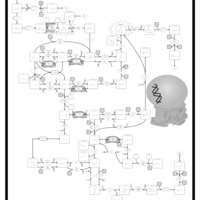 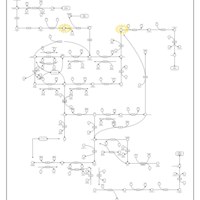 |
| Xanthine Dehydrogenase Deficiency (Xanthinuria) | 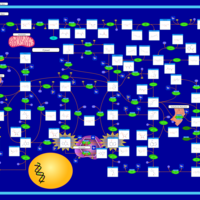 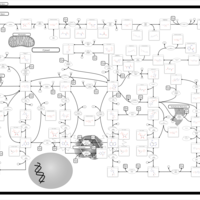  |
| Methionine Adenosyltransferase Deficiency |    |
| Glycine N-methyltransferase Deficiency | 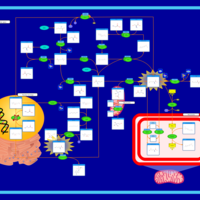 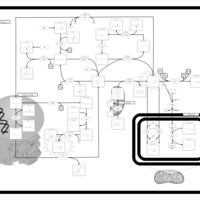 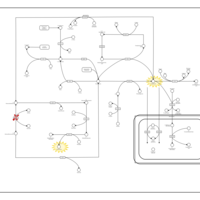 |
| Non Ketotic Hyperglycinemia | 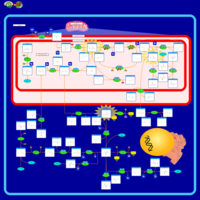 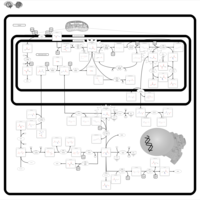 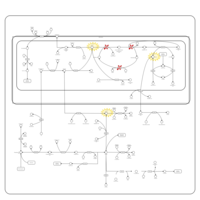 |
| Short Chain Acyl CoA Dehydrogenase Deficiency (SCAD Deficiency) | 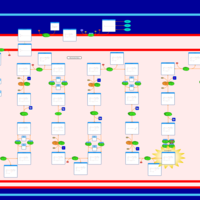 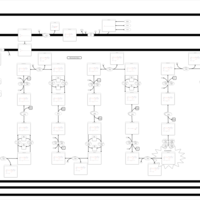 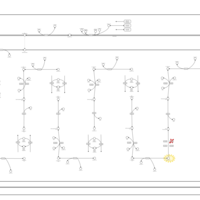 |
| Propionic Acidemia | 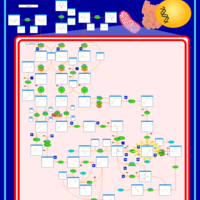 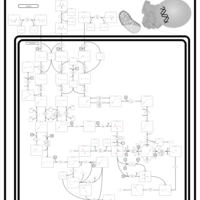 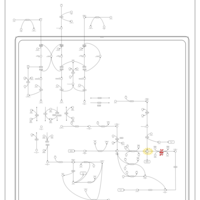 |
| 3-Methylcrotonyl Coa Carboxylase Deficiency Type I |  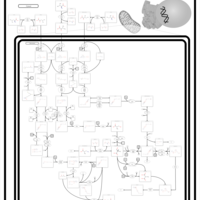 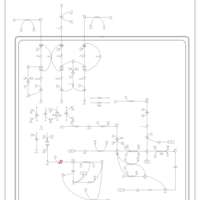 |
| Isovaleric Aciduria |    |
| Saccharopinuria/Hyperlysinemia II | 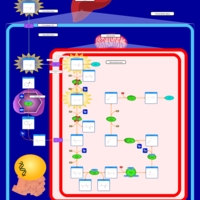 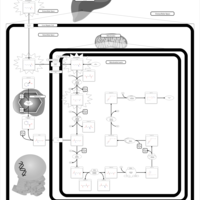 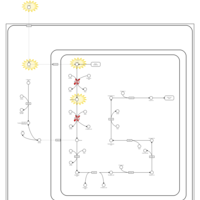 |
| Dimethylglycine Dehydrogenase Deficiency | 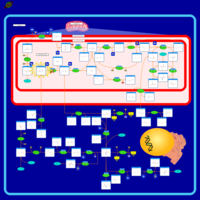 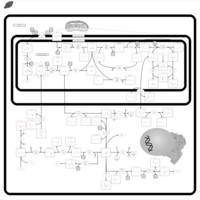  |
| 4-Hydroxybutyric Aciduria/Succinic Semialdehyde Dehydrogenase Deficiency | 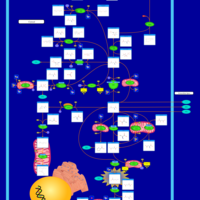 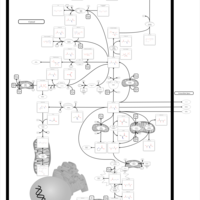 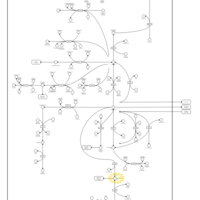 |
| Sarcosinemia | 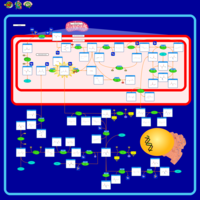 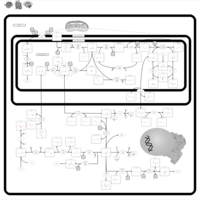 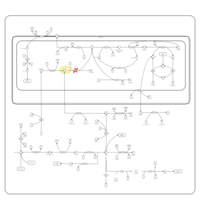 |
| Congenital Bile Acid Synthesis Defect Type II | 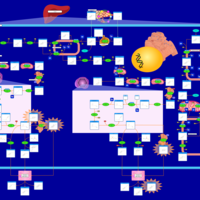 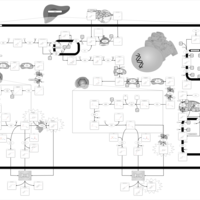 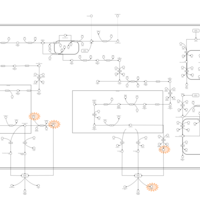 |
| Cerebrotendinous Xanthomatosis (CTX) |    |
| Zellweger Syndrome | 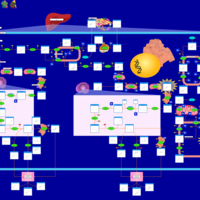 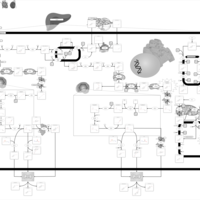 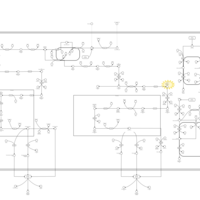 |
| Familial Hypercholanemia (FHCA) | 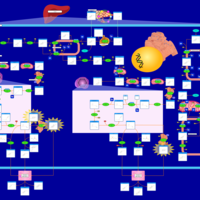 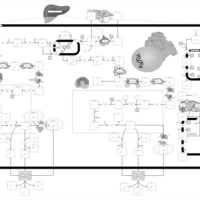 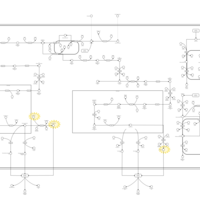 |
| Congenital Bile Acid Synthesis Defect Type III | 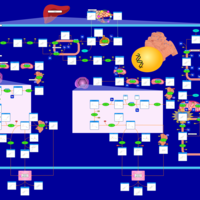 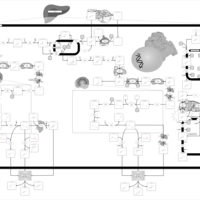 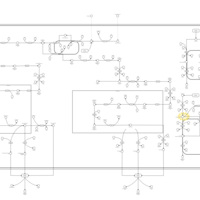 |
| Lysosomal Acid Lipase Deficiency (Wolman Disease) | 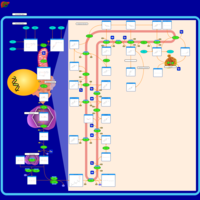 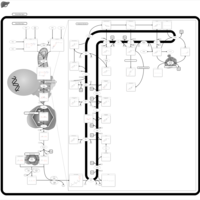 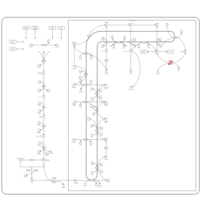 |
| Phenytoin (Antiarrhythmic) Action Pathway | 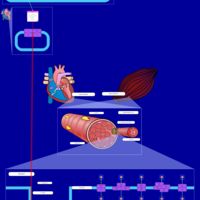 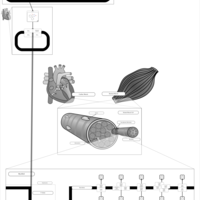  |
| Pyruvate Decarboxylase E1 Component Deficiency (PDHE1 Deficiency) | 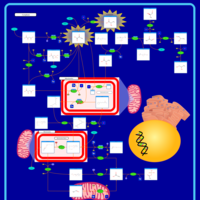 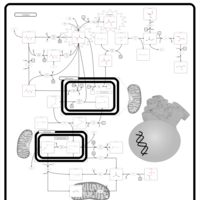 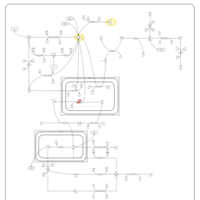 |
| Glutathione Synthetase Deficiency | 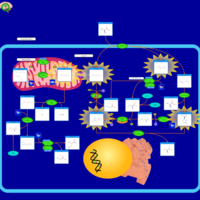 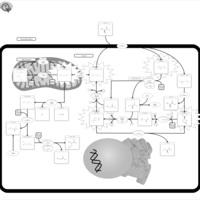 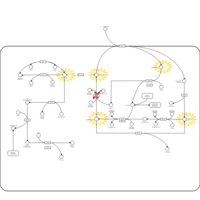 |
| Hyperinsulinism-Hyperammonemia Syndrome | 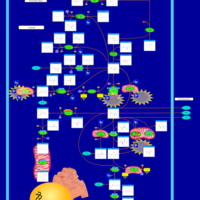 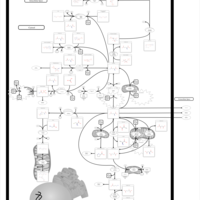 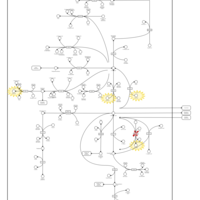 |
| Methylenetetrahydrofolate Reductase Deficiency (MTHFRD) |    |
| Hypermethioninemia |   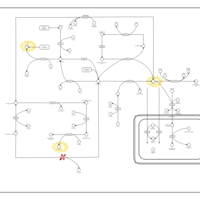 |
| Hereditary Coproporphyria (HCP) | 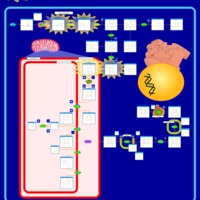 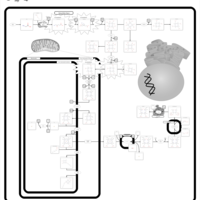 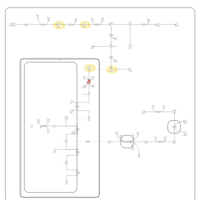 |
| Acute Intermittent Porphyria | 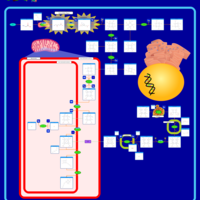 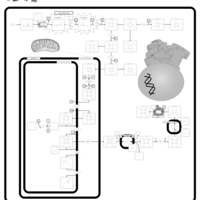  |
| Congenital Erythropoietic Porphyria (CEP) or Gunther Disease | 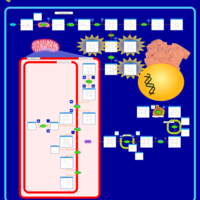 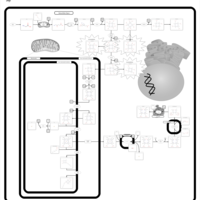 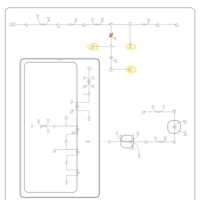 |
| Porphyria Variegata (PV) | 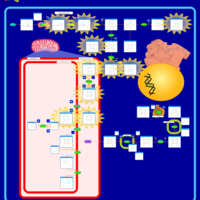 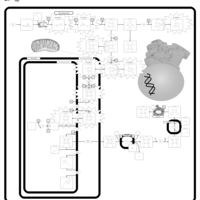 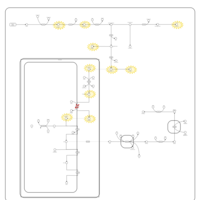 |
| GABA-Transaminase Deficiency |   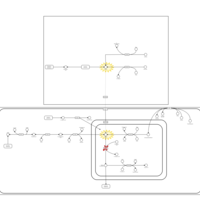 |
| Hyperprolinemia Type II | 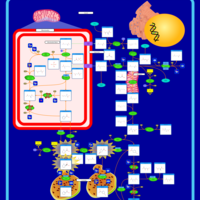 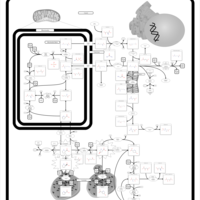 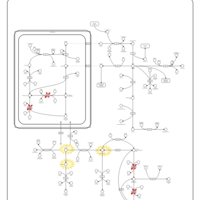 |
| Hyperprolinemia Type I | 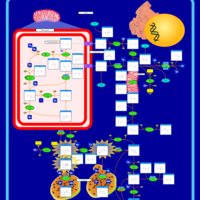 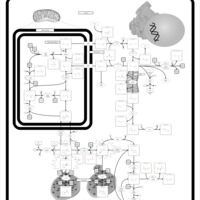 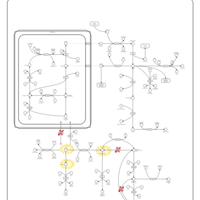 |
| Arginine: Glycine Amidinotransferase Deficiency (AGAT Deficiency) |  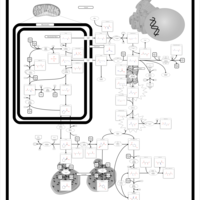 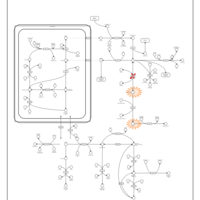 |
| Ornithine Aminotransferase Deficiency (OAT Deficiency) |    |
| Lesch-Nyhan Syndrome (LNS) |   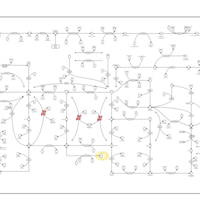 |
| Gout or Kelley-Seegmiller Syndrome | 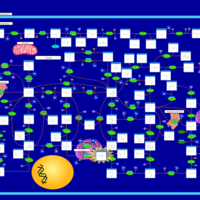 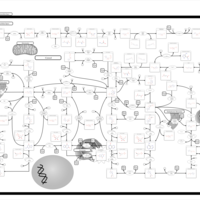 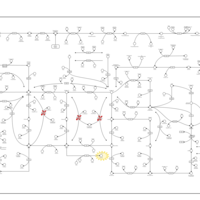 |
| Tyrosinemia Type 2 (or Richner-Hanhart syndrome) | 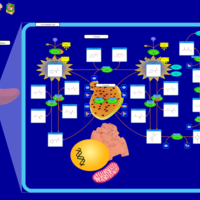 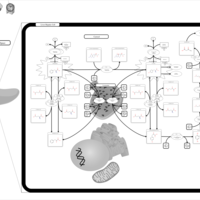 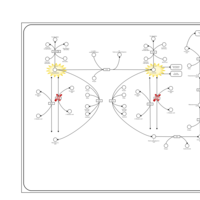 |
| Tyrosinemia Type 3 (TYRO3) | 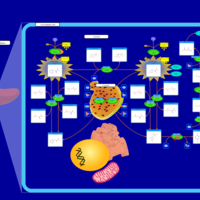 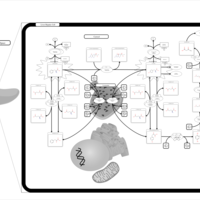  |
| Methylmalonate Semialdehyde Dehydrogenase Deficiency |    |
| Homocarnosinosis |    |
| Desmosterolosis | 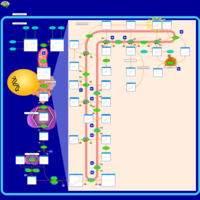 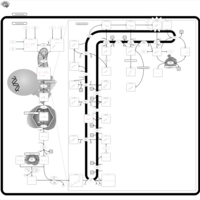 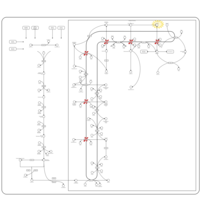 |
| CHILD Syndrome | 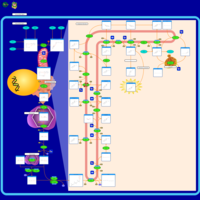 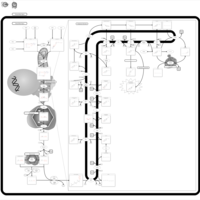 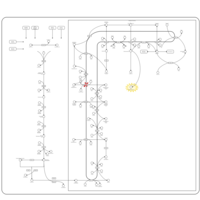 |
| Chondrodysplasia Punctata II, X Linked Dominant (CDPX2) |  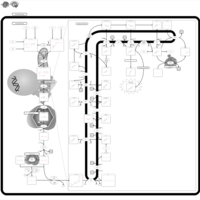 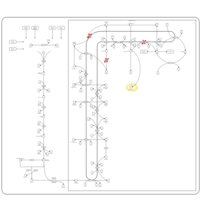 |
| Smith-Lemli-Opitz Syndrome (SLOS) |   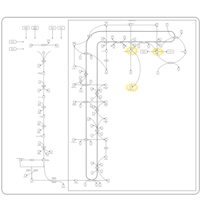 |
| Citalopram Action Pathway | 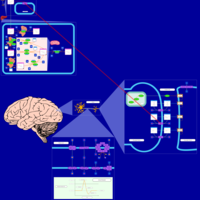 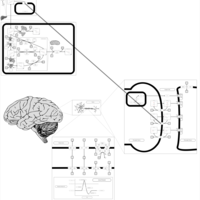  |
| Azathioprine Action Pathway | 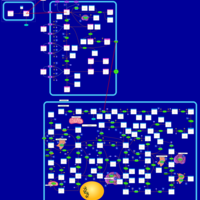 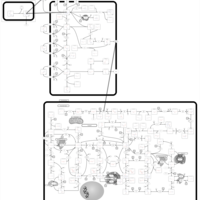 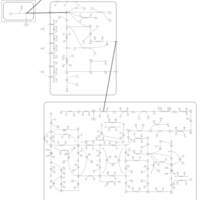 |
| Mercaptopurine Action Pathway |  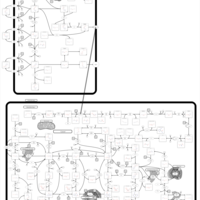 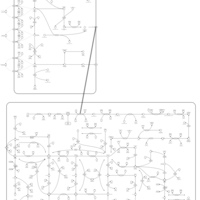 |
| Disulfiram Action Pathway | 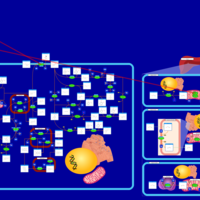   |
| Thioguanine Action Pathway | 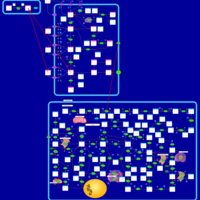   |
| Nicotine Action Pathway |    |
| Methotrexate Action Pathway | 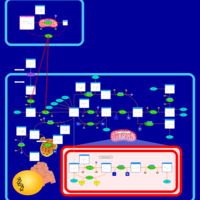 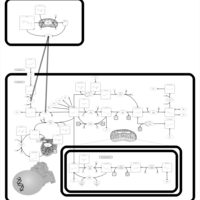 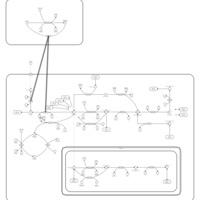 |
| Threonine and 2-Oxobutanoate Degradation | 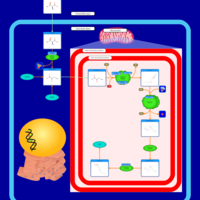 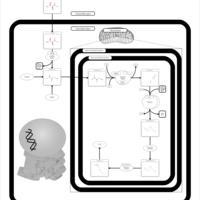 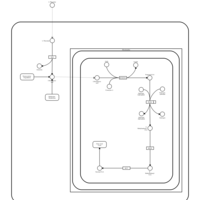 |
| Pyruvaldehyde Degradation | 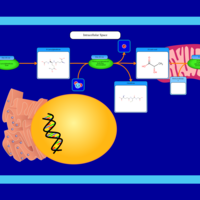 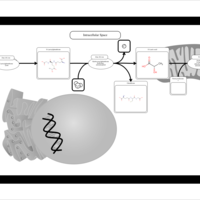 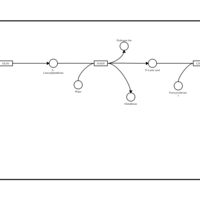 |
| Vitamin K Metabolism | 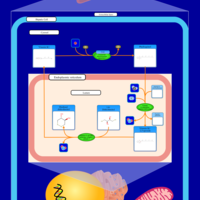 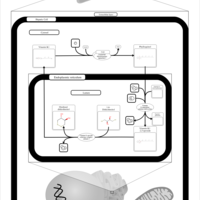 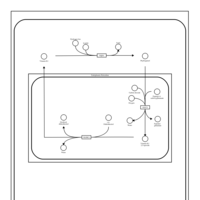 |
| Capecitabine Action Pathway | 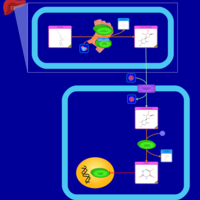 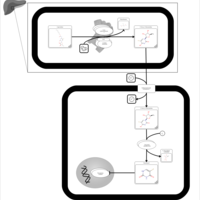  |
| Tamoxifen Action Pathway | 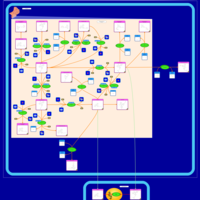 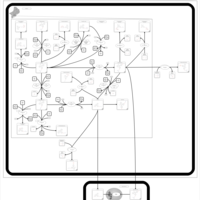 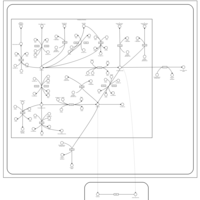 |
| Plasmalogen Synthesis | 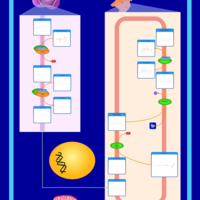 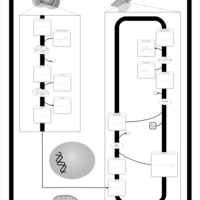 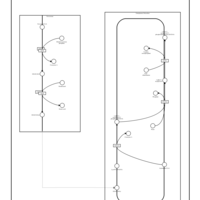 |
| Mitochondrial Beta-Oxidation of Short Chain Saturated Fatty Acids | 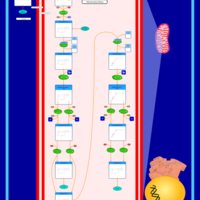 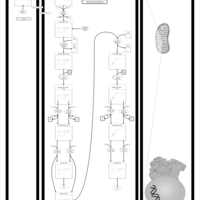  |
| Mitochondrial Beta-Oxidation of Medium Chain Saturated Fatty Acids | 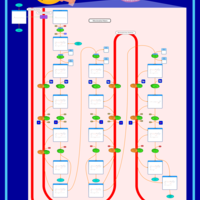 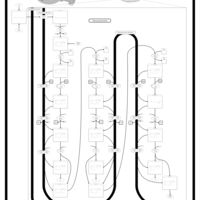 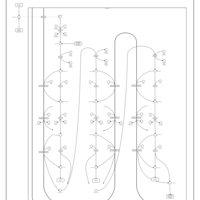 |
| Mitochondrial Beta-Oxidation of Long Chain Saturated Fatty Acids | 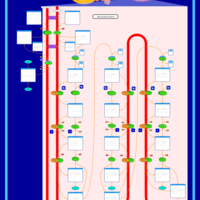 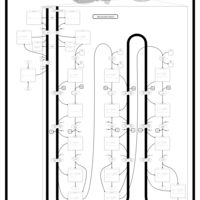 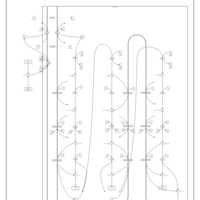 |
| Dimethylglycine Dehydrogenase Deficiency |    |
| Hyperglycinemia, non-ketotic |    |
| Ureidopropionase Deficiency |   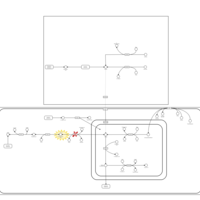 |
| Carnosinuria, carnosinemia |    |
| Tyrosinemia, transient, of the newborn |  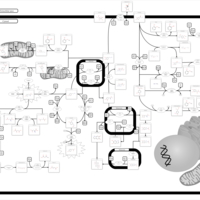 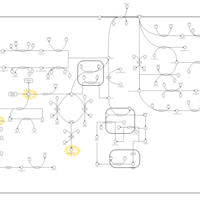 |
| Dopamine beta-hydroxylase deficiency |    |
| 5-oxoprolinase deficiency | 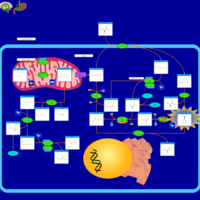 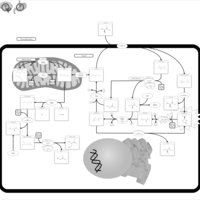 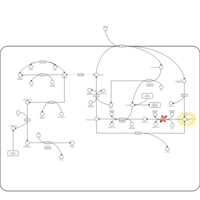 |
| Gamma-glutamyl-transpeptidase deficiency | 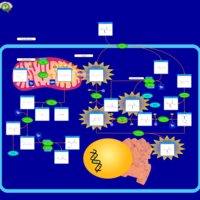 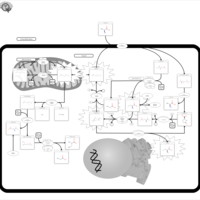 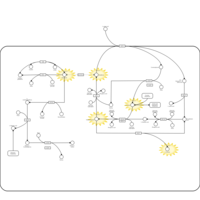 |
| Malonyl-coa decarboxylase deficiency | 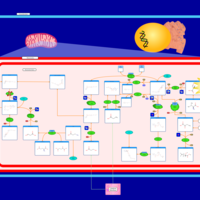 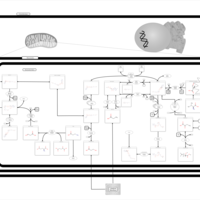 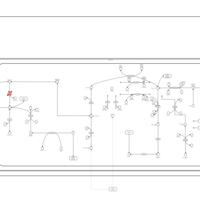 |
| Hypophosphatasia | 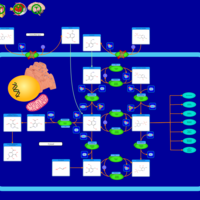 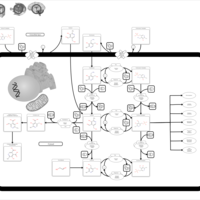 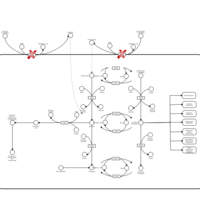 |
| Creatine deficiency, guanidinoacetate methyltransferase deficiency |    |
| Hyperornithinemia with gyrate atrophy (HOGA) |    |
| Hyperornithinemia-hyperammonemia-homocitrullinuria [HHH-syndrome] |    |
| L-arginine:glycine amidinotransferase deficiency |    |
| Cholesteryl ester storage disease |    |
| Hyper-IgD syndrome |    |
| Mevalonic aciduria |    |
| Wolman disease | 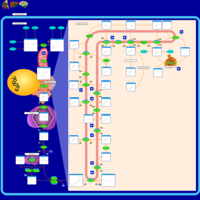 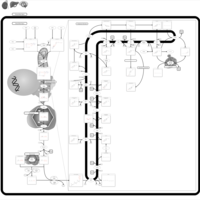 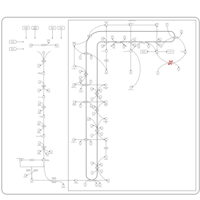 |
| Xanthinuria type I |  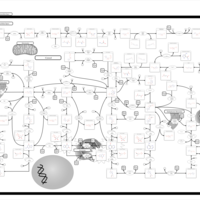 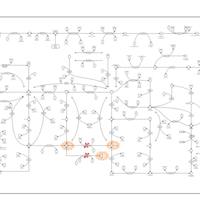 |
| Xanthinuria type II | 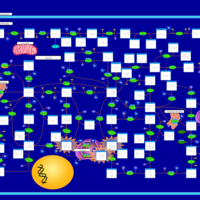   |
| 3-hydroxyisobutyric acid dehydrogenase deficiency |    |
| 3-hydroxyisobutyric aciduria |  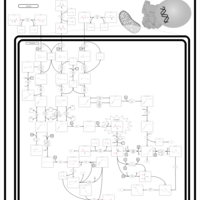 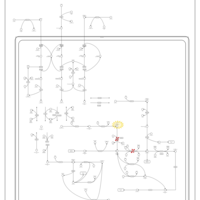 |
| Isobutyryl-coa dehydrogenase deficiency |    |
| Isovaleric acidemia |    |
| Hyperlysinemia I, Familial | 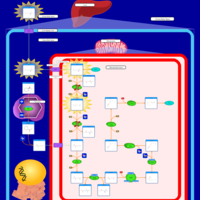 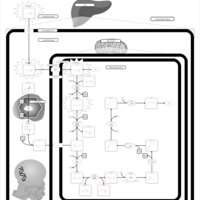 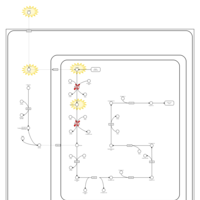 |
| Hyperlysinemia II or Saccharopinuria | 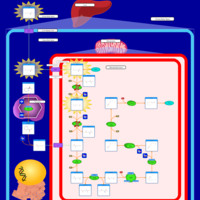 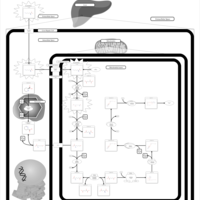  |
| D-glyceric acidura | 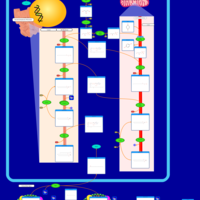 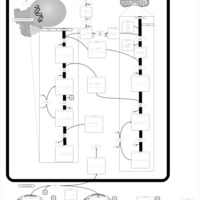 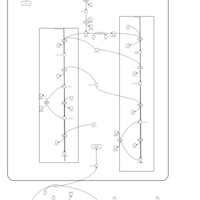 |
| Familial lipoprotein lipase deficiency | 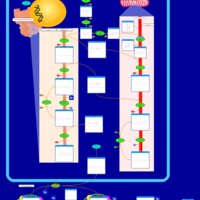  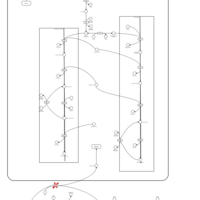 |
| Monoamine oxidase-a deficiency (MAO-A) |    |
| Adenine phosphoribosyltransferase deficiency (APRT) | 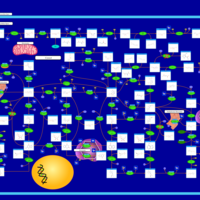 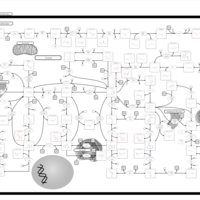 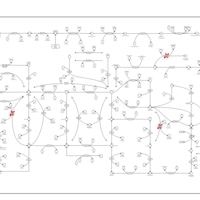 |
| Mitochondrial DNA depletion syndrome |    |
| Myoadenylate deaminase deficiency |    |
| Carnitine palmitoyl transferase deficiency (I) | 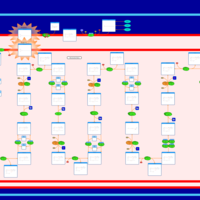 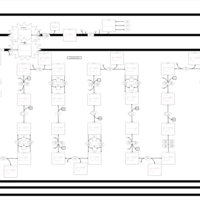 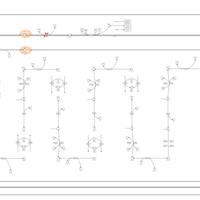 |
| Long chain acyl-CoA dehydrogenase deficiency (LCAD) |    |
| Very-long-chain acyl coa dehydrogenase deficiency (VLCAD) | 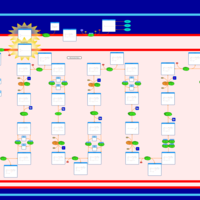 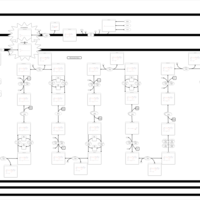 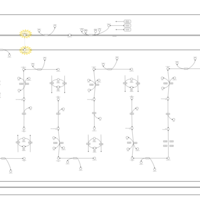 |
| Carnitine palmitoyl transferase deficiency (II) |    |
| Medium chain acyl-coa dehydrogenase deficiency (MCAD) |    |
| Methylenetetrahydrofolate Reductase Deficiency (MTHFRD) | 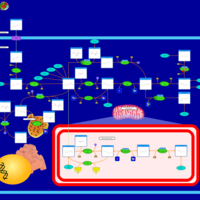 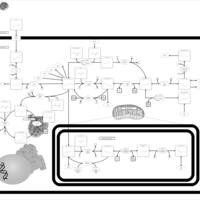 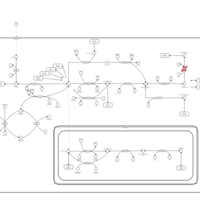 |
| Trifunctional protein deficiency | 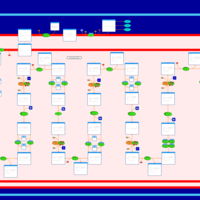 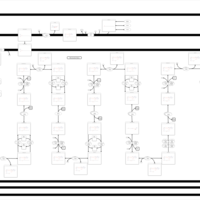 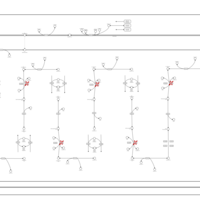 |
| Congenital lactic acidosis | 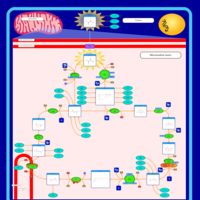 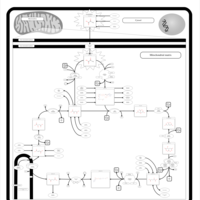 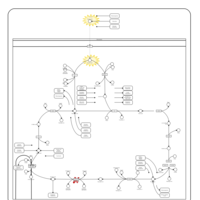 |
| Fumarase deficiency | 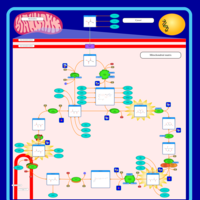 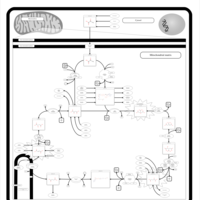 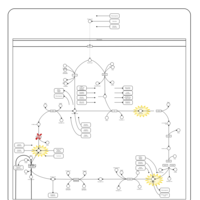 |
| Mitochondrial complex II deficiency |  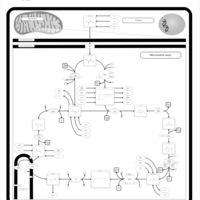 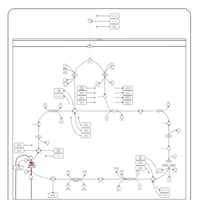 |
| 2-ketoglutarate dehydrogenase complex deficiency | 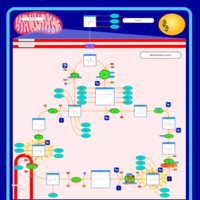 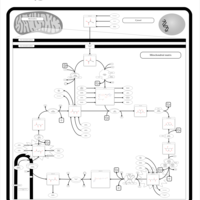 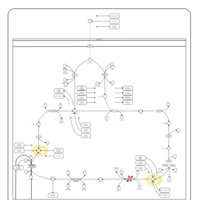 |
| Pyruvate dehydrogenase deficiency (E3) | 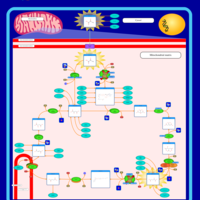 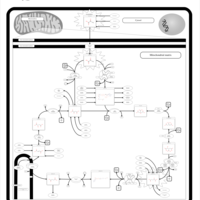 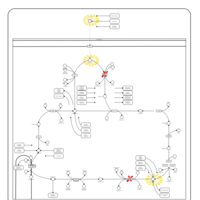 |
| Pyruvate dehydrogenase deficiency (E2) | 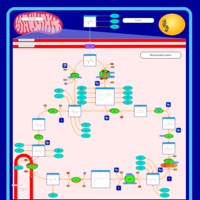 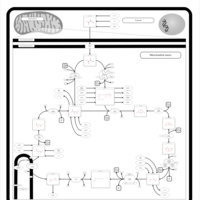 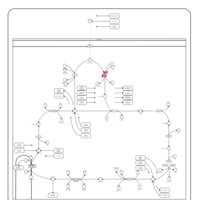 |
| Primary hyperoxaluria II, PH2 | 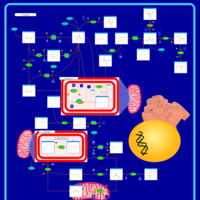 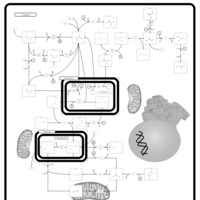 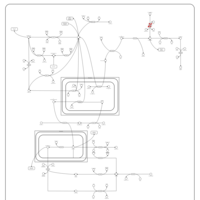 |
| Pyruvate kinase deficiency | 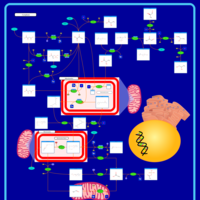 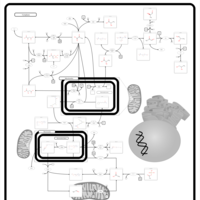 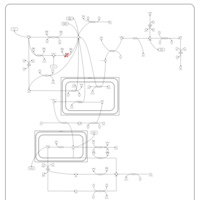 |
| Succinic semialdehyde dehydrogenase deficiency | 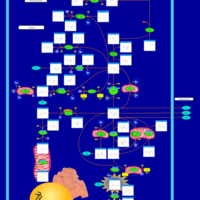 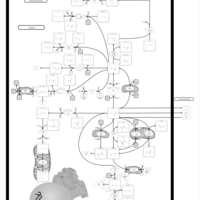 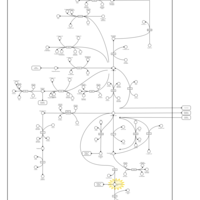 |
| Short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (SCHAD) | 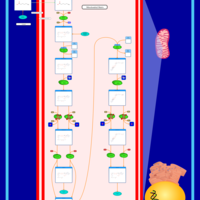 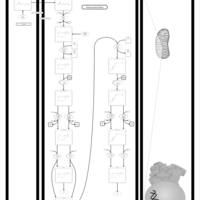 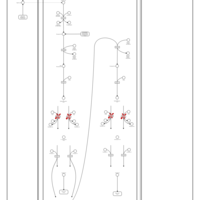 |
| Homocystinuria-megaloblastic anemia due to defect in cobalamin metabolism, cblG complementation type |    |
| Pyridoxine dependency with seizures |  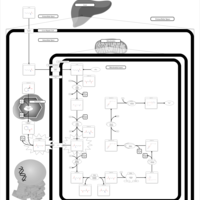 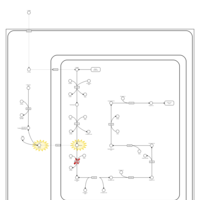 |
| Tamoxifen Metabolism Pathway | 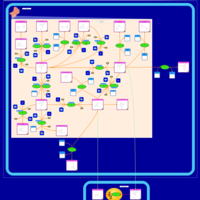 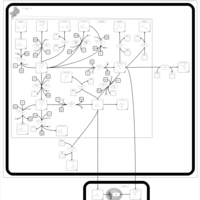  |
| Capecitabine Metabolism Pathway | 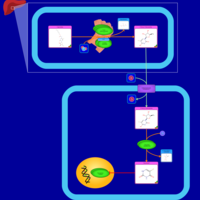  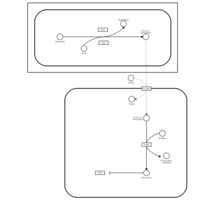 |
| Mercaptopurine Metabolism Pathway | 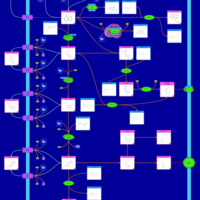 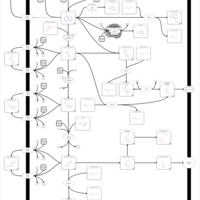 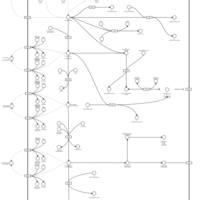 |
| Citalopram Metabolism Pathway | 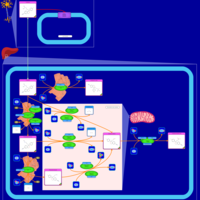 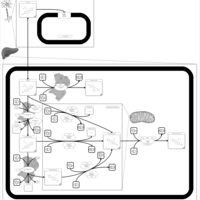 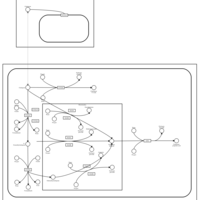 |
| Nicotine Metabolism Pathway | 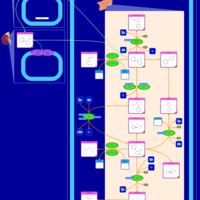 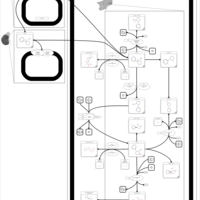 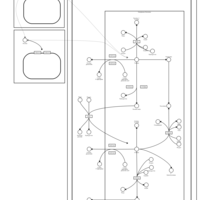 |
| Valproic Acid Metabolism Pathway | 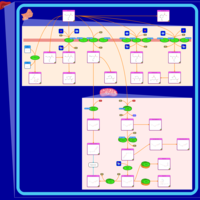 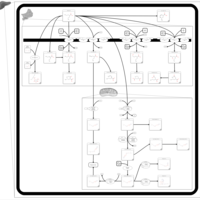 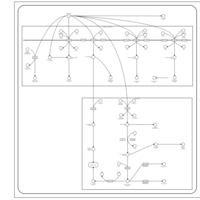 |
| Doxorubicin Metabolism Pathway | 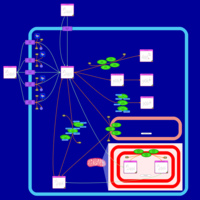  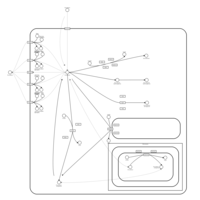 |
| Warburg Effect | 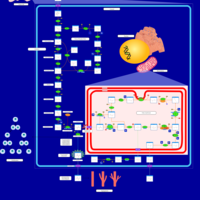 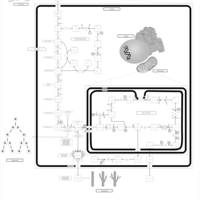 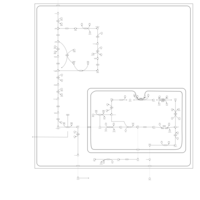 |
| Thyroid hormone synthesis |  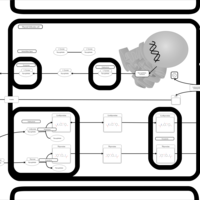 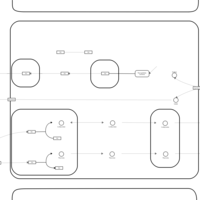 |
| 2-aminoadipic 2-oxoadipic aciduria | 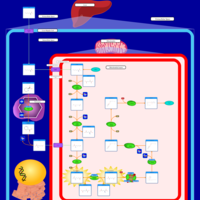  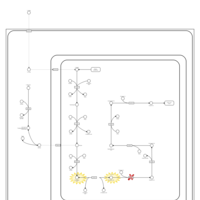 |
| 27-Hydroxylase Deficiency | 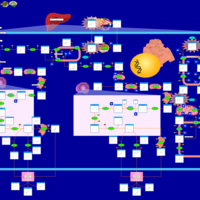 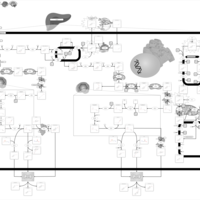 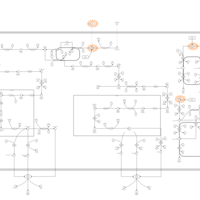 |
| 3-Phosphoglycerate dehydrogenase deficiency | 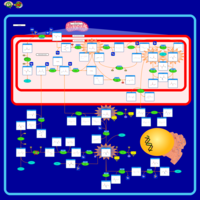 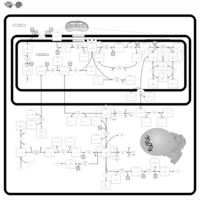 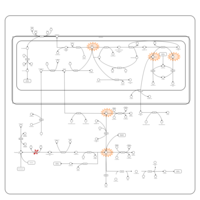 |
| Folate malabsorption, hereditary | 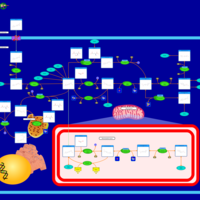 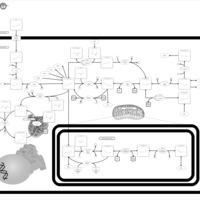 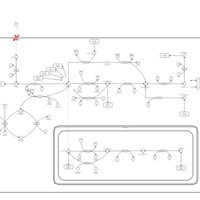 |
| The oncogenic action of 2-hydroxyglutarate | 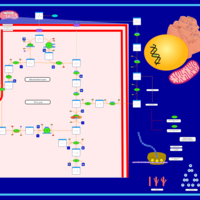 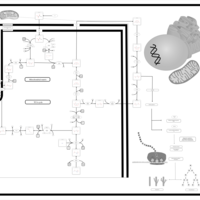 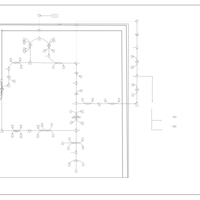 |
| The Oncogenic Action of Succinate | 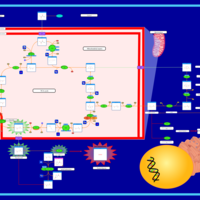  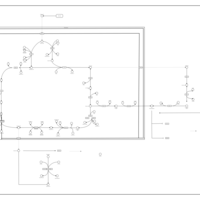 |
| The Oncogenic Action of Fumarate | 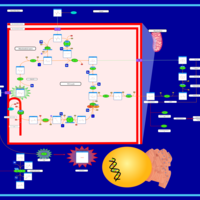 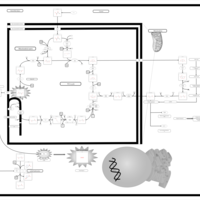  |
| Glutaminolysis and Cancer | 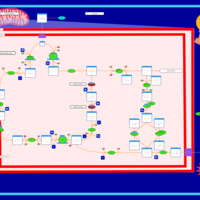 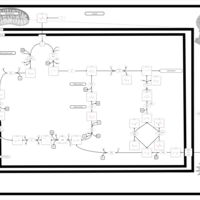 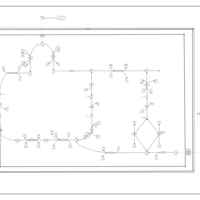 |
| Sarcosine Oncometabolite Pathway |  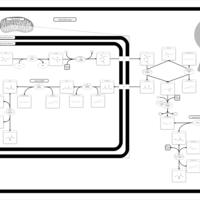  |
| The oncogenic action of L-2-hydroxyglutarate in Hydroxygluaricaciduria | 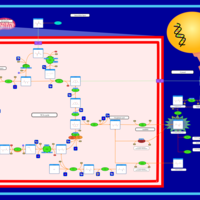 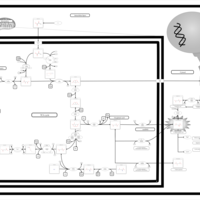 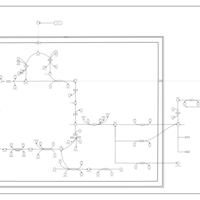 |
| The oncogenic action of D-2-hydroxyglutarate in Hydroxygluaricaciduria | 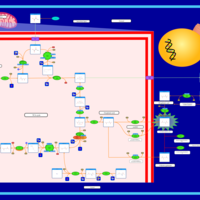 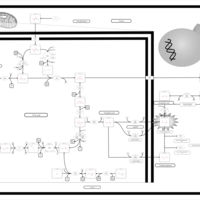 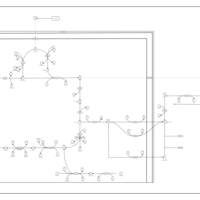 |
