| Valine, leucine and isoleucine degradation |  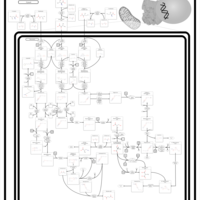 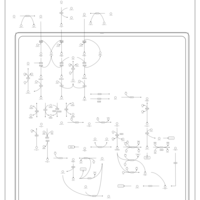 |
| Pyruvate metabolism |  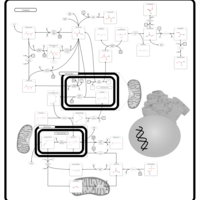  |
| Lysine degradation |  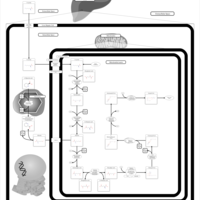 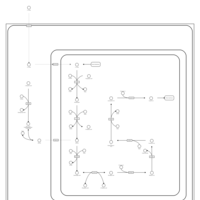 |
| Propanoate metabolism | 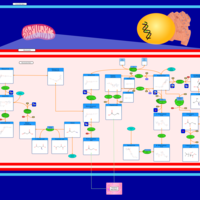 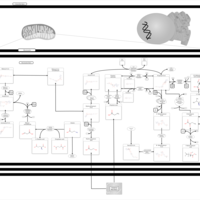 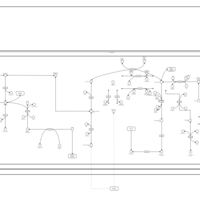 |
| Oxidation of Branched Chain Fatty Acids | 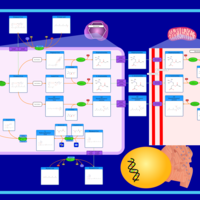 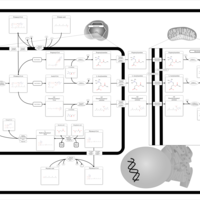 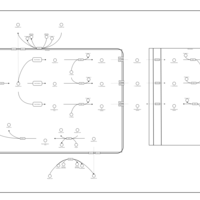 |
| Citric Acid Cycle | 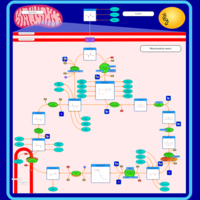 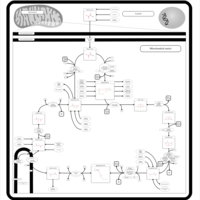 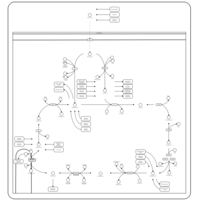 |
| Thiamine Metabolism | 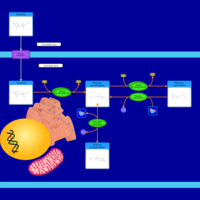 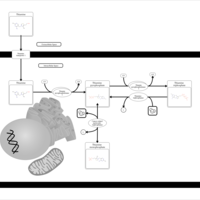 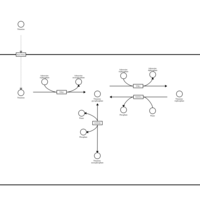 |
| 2-Methyl-3-Hydroxybutryl CoA Dehydrogenase Deficiency | 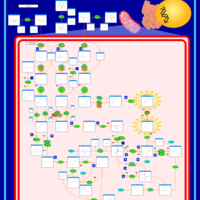 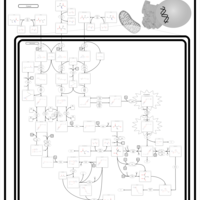 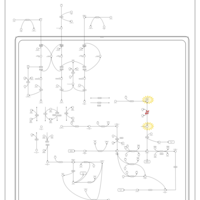 |
| 3-Hydroxy-3-Methylglutaryl-CoA Lyase Deficiency |  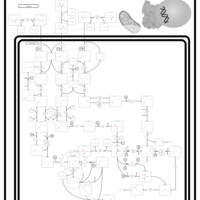 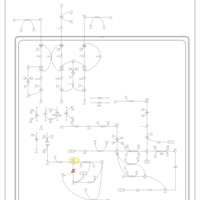 |
| 3-Methylglutaconic Aciduria Type I |  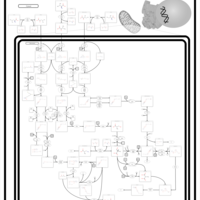 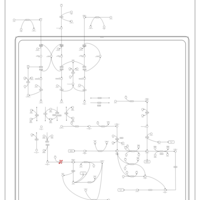 |
| 3-Methylglutaconic Aciduria Type III | 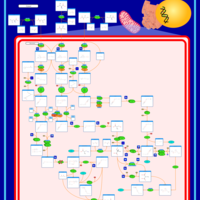 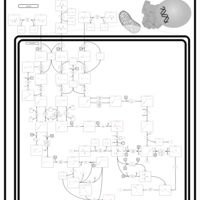 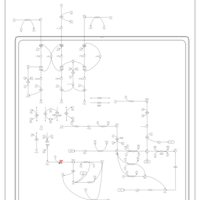 |
| 3-Methylglutaconic Aciduria Type IV |  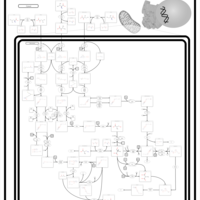 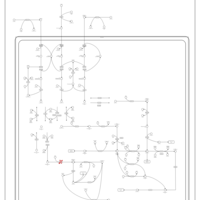 |
| Beta-Ketothiolase Deficiency | 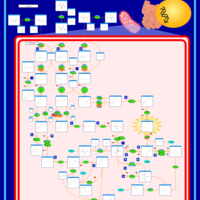 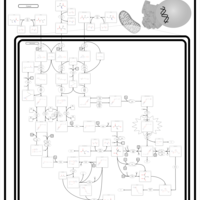 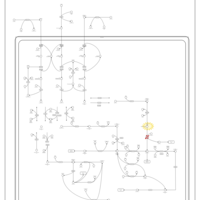 |
| Glutaric Aciduria Type I | 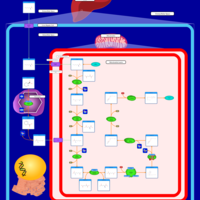 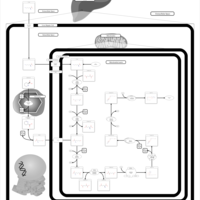 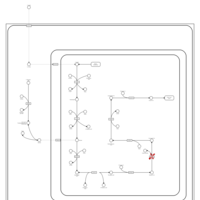 |
| Leigh Syndrome | 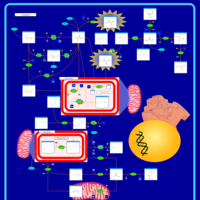 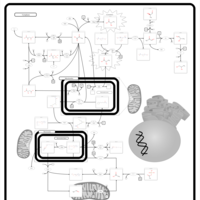 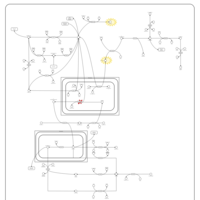 |
| Malonic Aciduria | 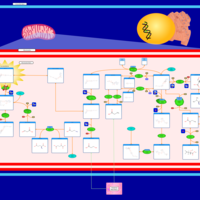  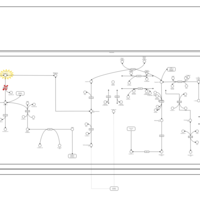 |
| Maple Syrup Urine Disease |    |
| Methylmalonic Aciduria |    |
| Methylmalonic Aciduria Due to Cobalamin-Related Disorders | 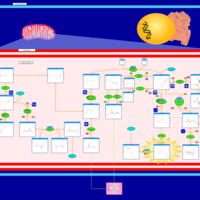 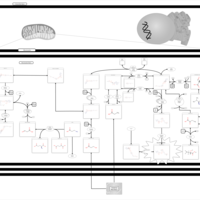 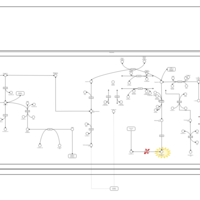 |
| Pyruvate Dehydrogenase Complex Deficiency |   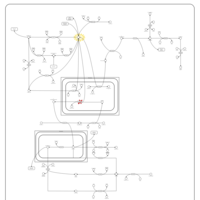 |
| Propionic Acidemia | 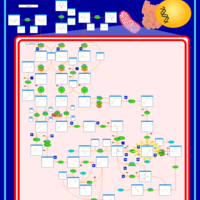 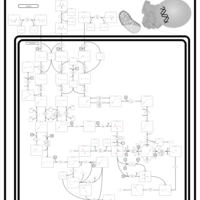 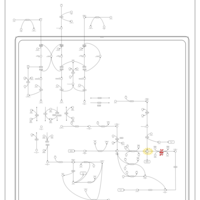 |
| 3-Methylcrotonyl Coa Carboxylase Deficiency Type I | 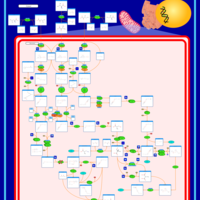 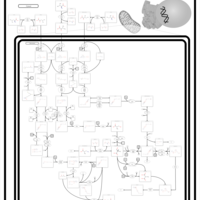 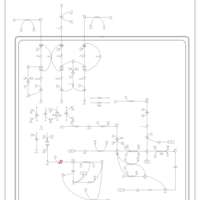 |
| Isovaleric Aciduria |    |
| Saccharopinuria/Hyperlysinemia II | 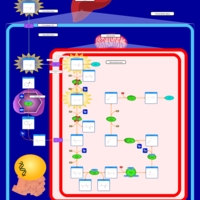 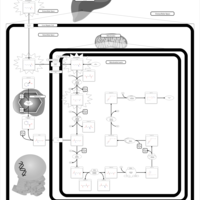 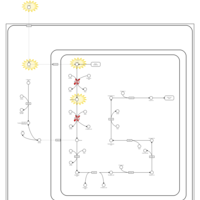 |
| Pyruvate Decarboxylase E1 Component Deficiency (PDHE1 Deficiency) | 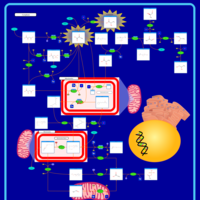 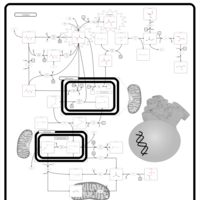 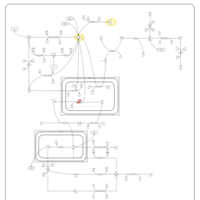 |
| Methylmalonate Semialdehyde Dehydrogenase Deficiency |    |
| Phytanic Acid Peroxisomal Oxidation | 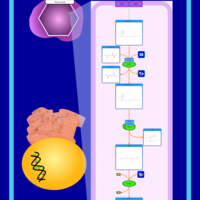 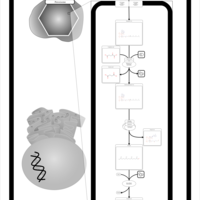 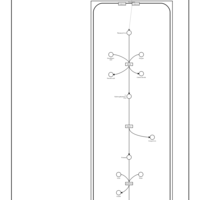 |
| Refsum Disease | 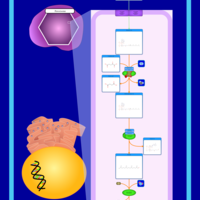 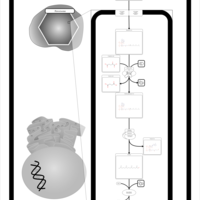 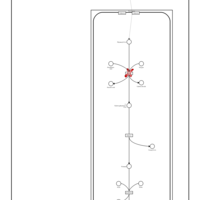 |
| Threonine and 2-Oxobutanoate Degradation | 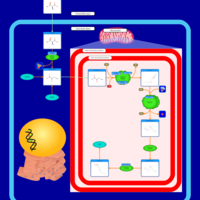 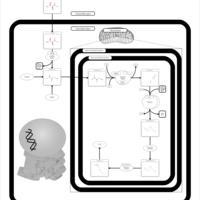 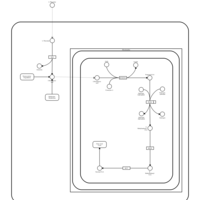 |
| Transfer of Acetyl Groups into Mitochondria | 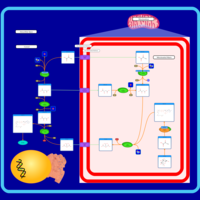 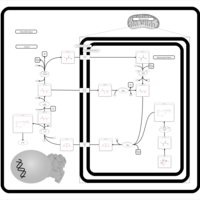 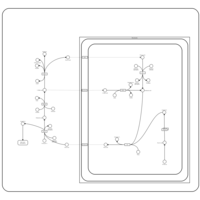 |
| Malonyl-coa decarboxylase deficiency | 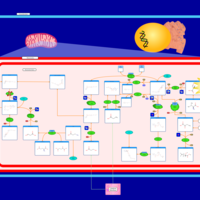 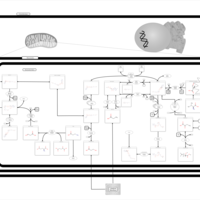 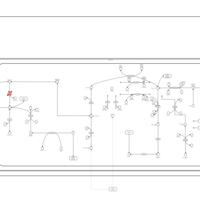 |
| 3-hydroxyisobutyric acid dehydrogenase deficiency |  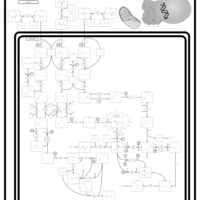 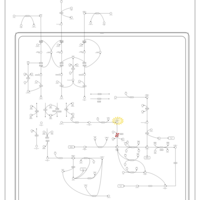 |
| 3-hydroxyisobutyric aciduria |  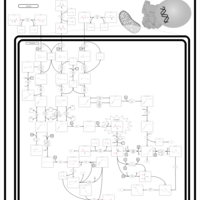 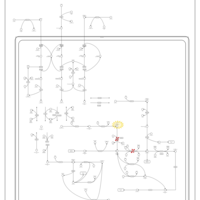 |
| Isobutyryl-coa dehydrogenase deficiency |    |
| Isovaleric acidemia |    |
| Hyperlysinemia I, Familial | 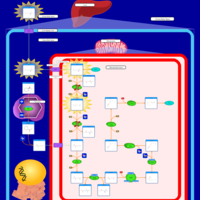 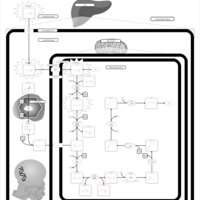 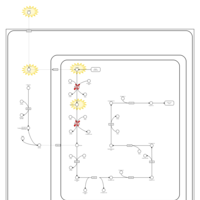 |
| Hyperlysinemia II or Saccharopinuria | 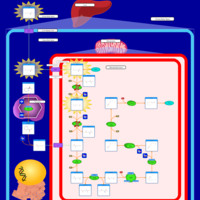 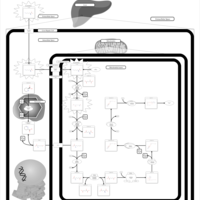  |
| Congenital lactic acidosis | 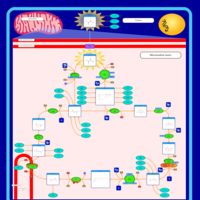 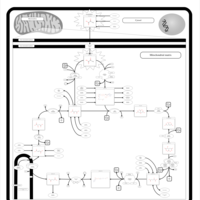 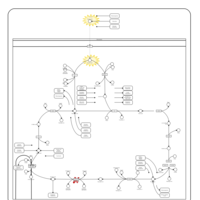 |
| Fumarase deficiency |  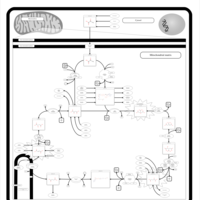 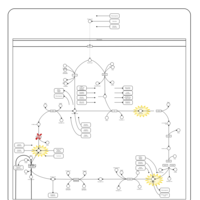 |
| Mitochondrial complex II deficiency | 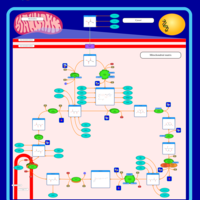 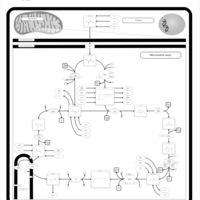 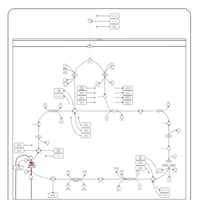 |
| 2-ketoglutarate dehydrogenase complex deficiency | 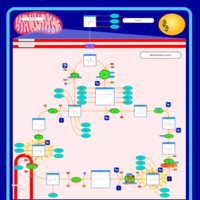 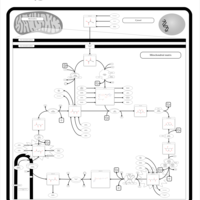 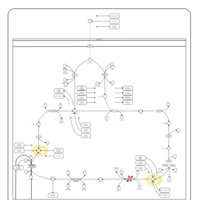 |
| Pyruvate dehydrogenase deficiency (E3) |  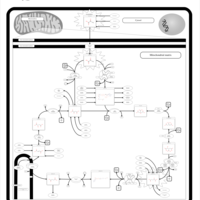 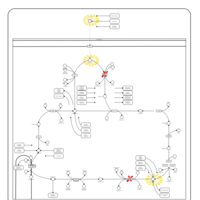 |
| Pyruvate dehydrogenase deficiency (E2) |  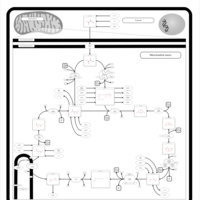 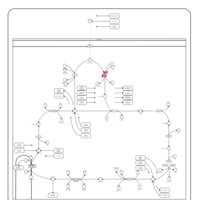 |
| Primary hyperoxaluria II, PH2 | 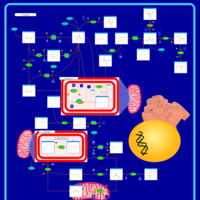 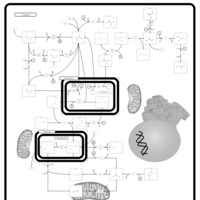  |
| Pyruvate kinase deficiency | 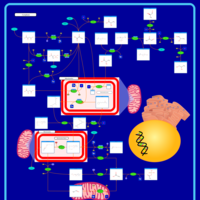 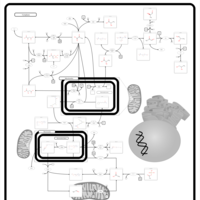 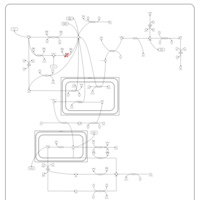 |
| Pyridoxine dependency with seizures |  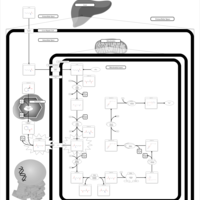 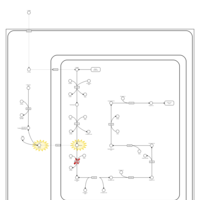 |
| Warburg Effect | 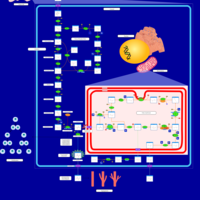   |
| 2-aminoadipic 2-oxoadipic aciduria | 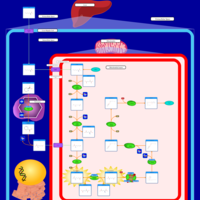  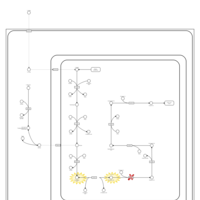 |
| The oncogenic action of 2-hydroxyglutarate | 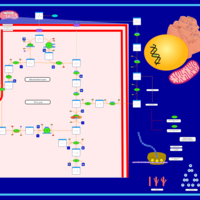 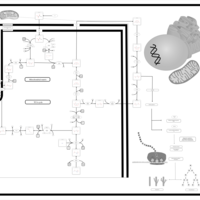 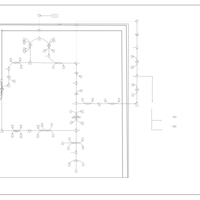 |
| The Oncogenic Action of Succinate | 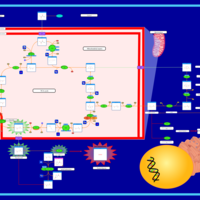  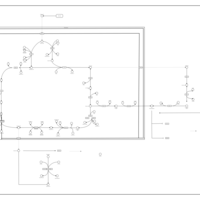 |
| The Oncogenic Action of Fumarate | 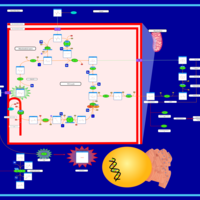 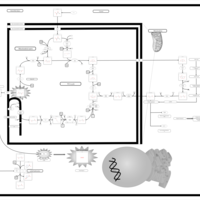  |
| Glutaminolysis and Cancer | 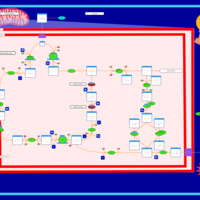 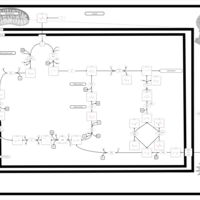 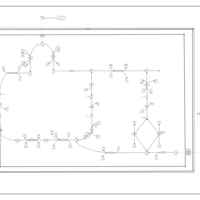 |
| The oncogenic action of L-2-hydroxyglutarate in Hydroxygluaricaciduria | 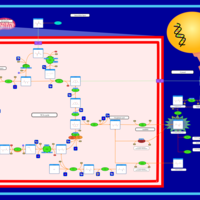 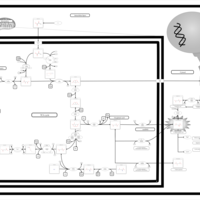 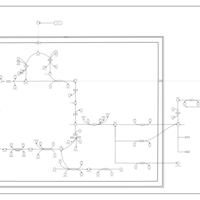 |
| The oncogenic action of D-2-hydroxyglutarate in Hydroxygluaricaciduria | 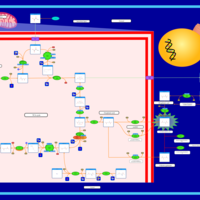 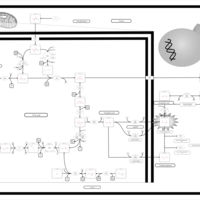 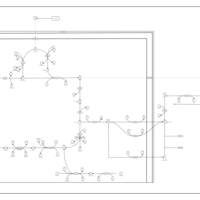 |
