| Alanine, aspartate and glutamate metabolism | 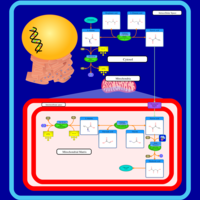   |
| Selenocompound metabolism |  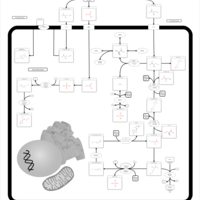  |
| Nitrogen metabolism |  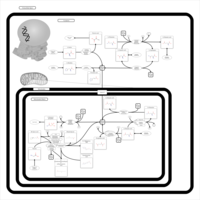 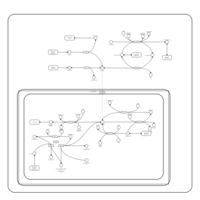 |
| Arginine and proline metabolism | 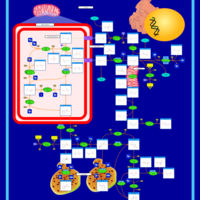 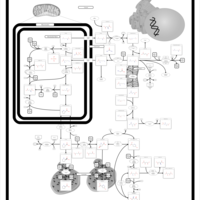 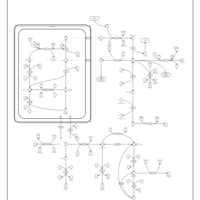 |
| beta-Alanine metabolism | 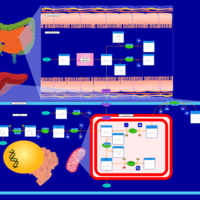 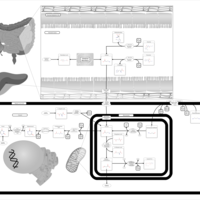 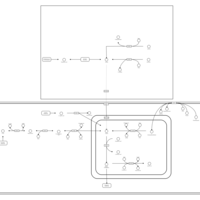 |
| Tyrosine metabolism | 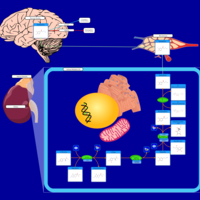   |
| One carbon pool by folate |   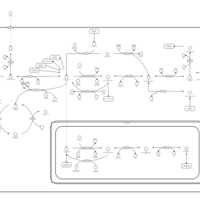 |
| Lysine degradation | 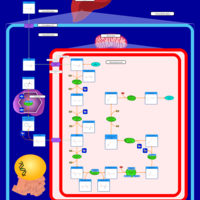 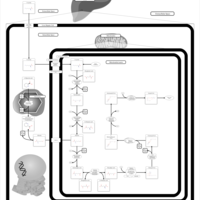 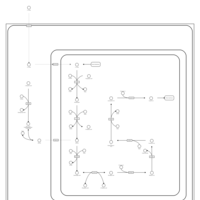 |
| Tyrosine metabolism | 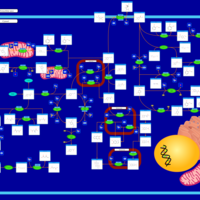 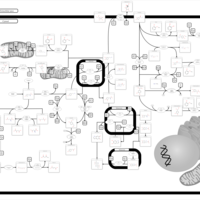 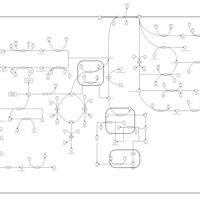 |
| Starch and sucrose metabolism | 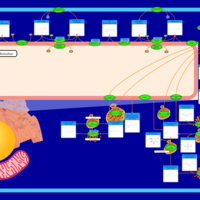 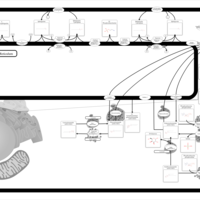 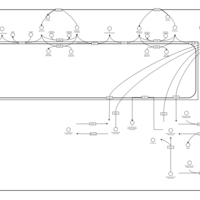 |
| Glycine, serine and threonine metabolism | 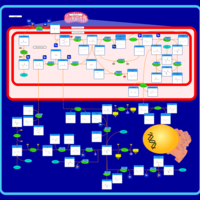 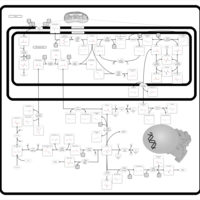 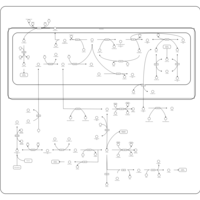 |
| Tryptophan metabolism |  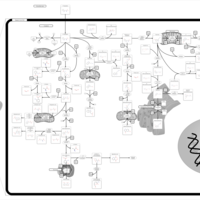 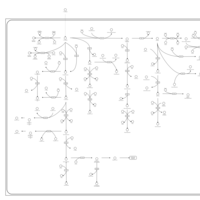 |
| Valine, leucine and isoleucine degradation |  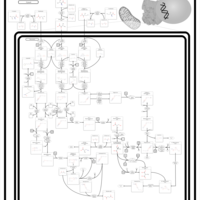 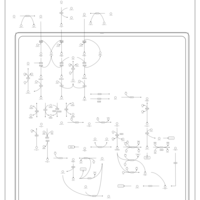 |
| Propanoate metabolism | 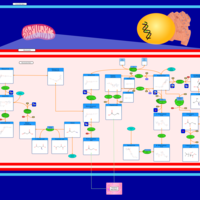 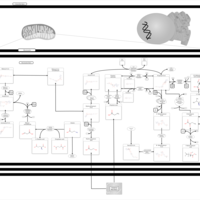 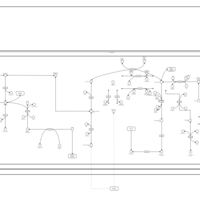 |
| Citrullinemia Type I | 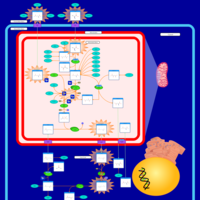 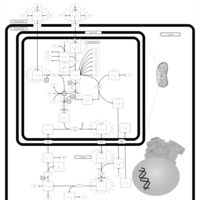 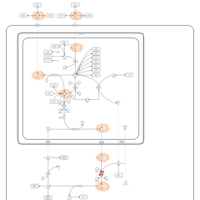 |
| Carbamoyl Phosphate Synthetase Deficiency | 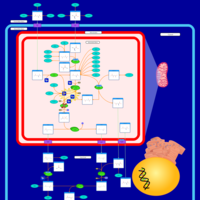 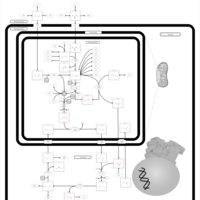 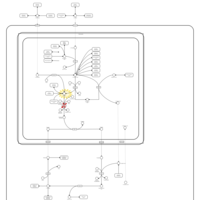 |
| Argininosuccinic Aciduria | 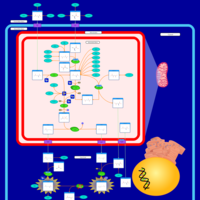  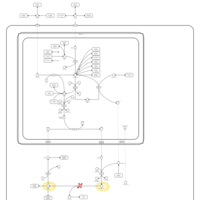 |
| Phenylalanine and Tyrosine Metabolism | 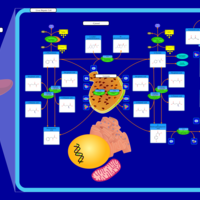 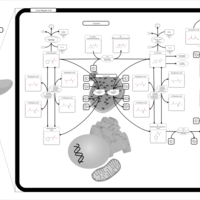 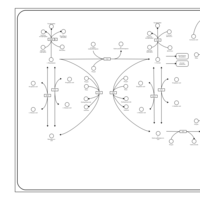 |
| Cysteine Metabolism | 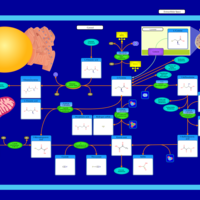 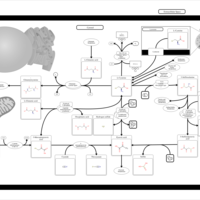 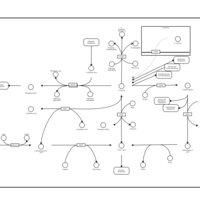 |
| Vitamin B6 Metabolism | 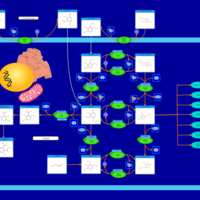 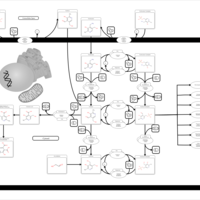 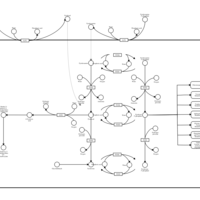 |
| Taurine and Hypotaurine Metabolism |    |
| Porphyrin Metabolism |    |
| Methionine Metabolism |  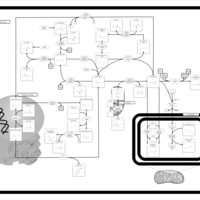 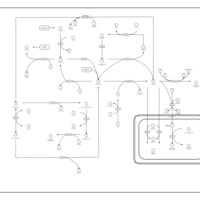 |
| Histidine metabolism | 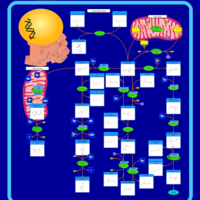 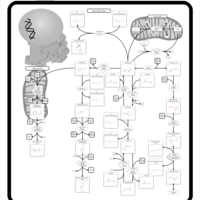 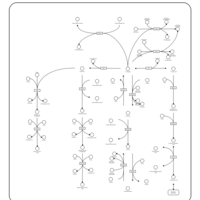 |
| Starch and Sucrose Metabolism | 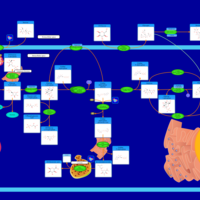 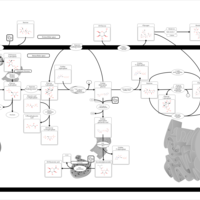 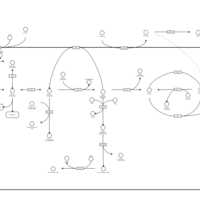 |
| Urea Cycle | 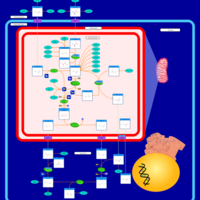 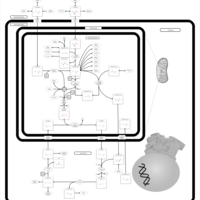 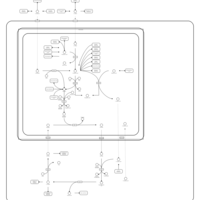 |
| Aspartate Metabolism | 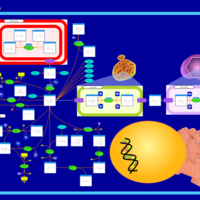 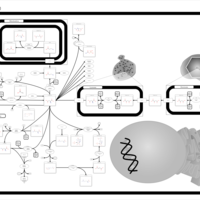 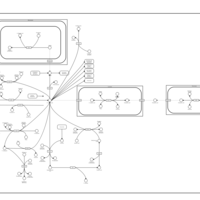 |
| Glutamate Metabolism | 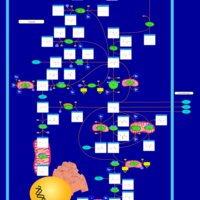 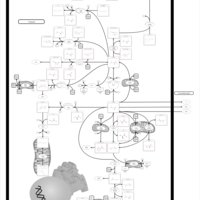  |
| Glucose-Alanine Cycle | 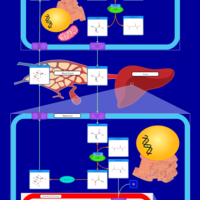 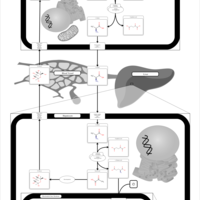  |
| Malate-Aspartate Shuttle |  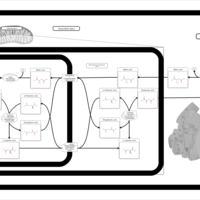 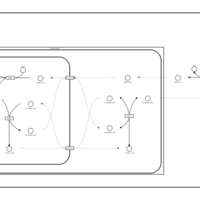 |
| 2-Hydroxyglutric Aciduria (D And L Form) | 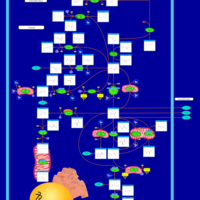 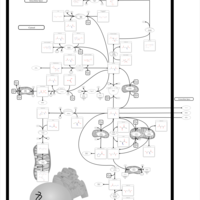 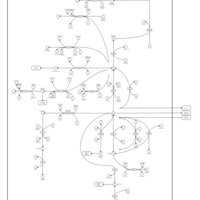 |
| 2-Methyl-3-Hydroxybutryl CoA Dehydrogenase Deficiency |  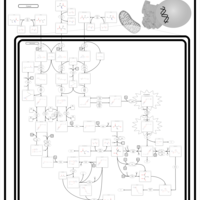 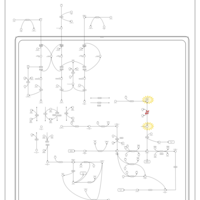 |
| 3-Hydroxy-3-Methylglutaryl-CoA Lyase Deficiency | 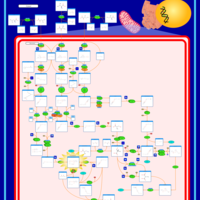 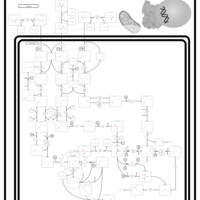 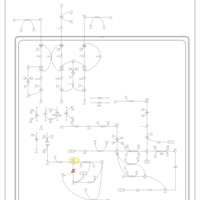 |
| 3-Methylglutaconic Aciduria Type I |  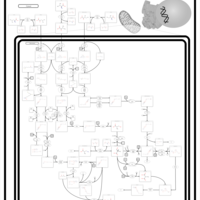 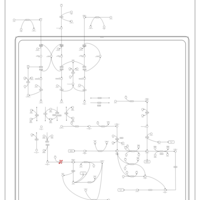 |
| 3-Methylglutaconic Aciduria Type III |  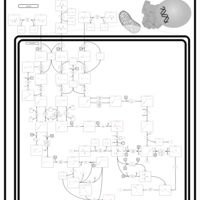 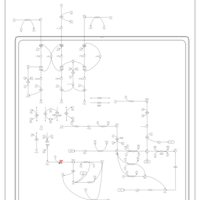 |
| 3-Methylglutaconic Aciduria Type IV | 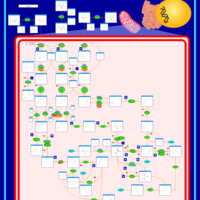 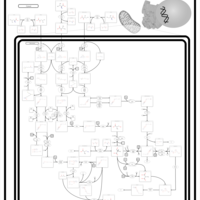 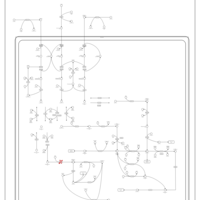 |
| Alkaptonuria |  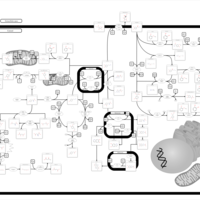 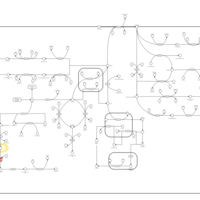 |
| Aromatic L-Aminoacid Decarboxylase Deficiency | 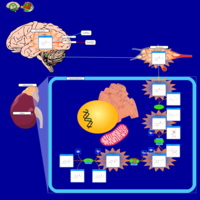 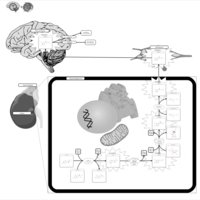  |
| Beta-Ketothiolase Deficiency |    |
| Canavan Disease | 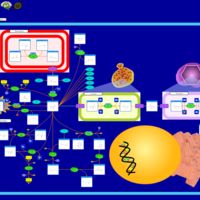 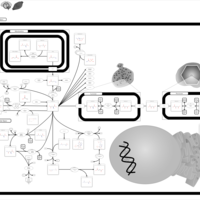 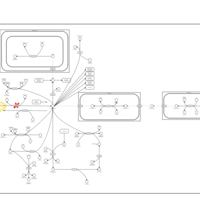 |
| Cystathionine Beta-Synthase Deficiency | 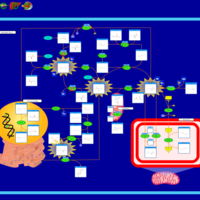 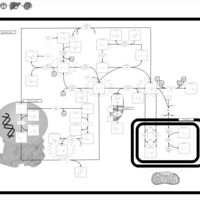 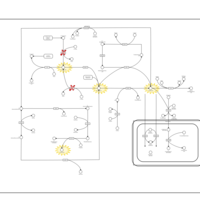 |
| Dihydropyrimidine Dehydrogenase Deficiency (DHPD) | 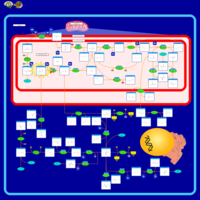 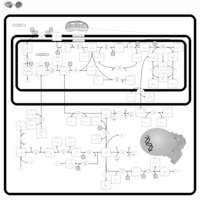  |
| Glutaric Aciduria Type I |    |
| Guanidinoacetate Methyltransferase Deficiency (GAMT Deficiency) |    |
| Hawkinsinuria | 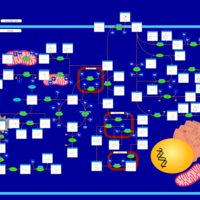  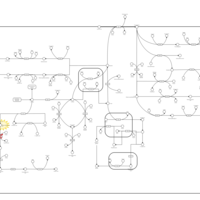 |
| Histidinemia | 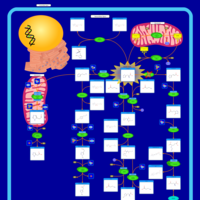 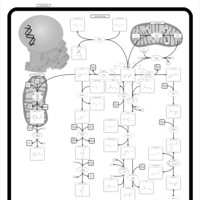  |
| Hypoacetylaspartia |    |
| Malonic Aciduria |   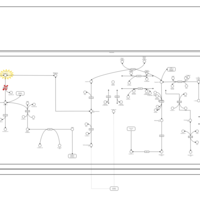 |
| Maple Syrup Urine Disease |    |
| Methylmalonic Aciduria |    |
| Methylmalonic Aciduria Due to Cobalamin-Related Disorders |   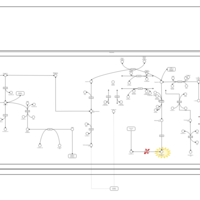 |
| Ornithine Transcarbamylase Deficiency (OTC Deficiency) | 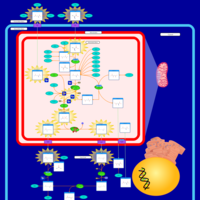   |
| Phenylketonuria | 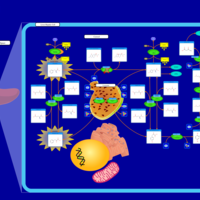 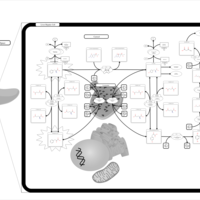 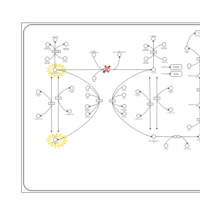 |
| Prolidase Deficiency (PD) | 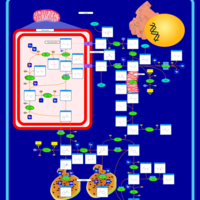 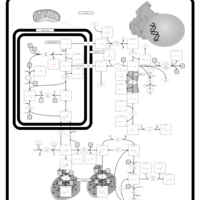 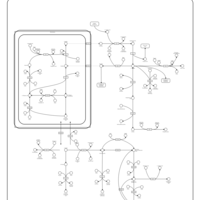 |
| Prolinemia Type II |   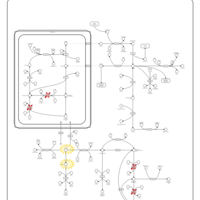 |
| S-Adenosylhomocysteine (SAH) Hydrolase Deficiency |   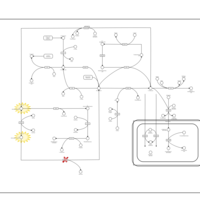 |
| Tyrosinemia Type I | 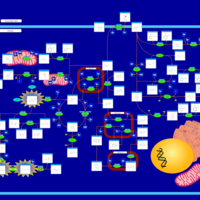 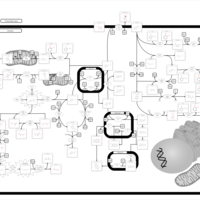 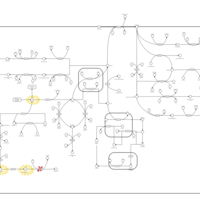 |
| Methionine Adenosyltransferase Deficiency |    |
| Glycine N-methyltransferase Deficiency | 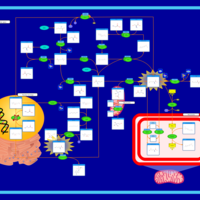 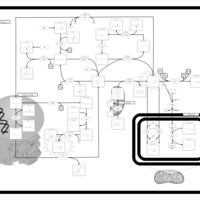 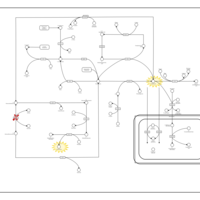 |
| Non Ketotic Hyperglycinemia | 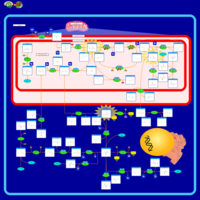 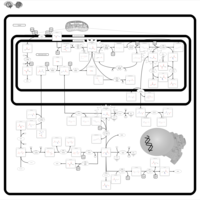 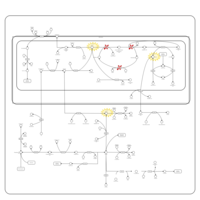 |
| Propionic Acidemia |   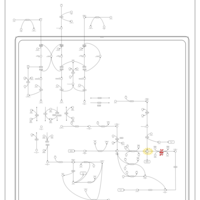 |
| 3-Methylcrotonyl Coa Carboxylase Deficiency Type I |  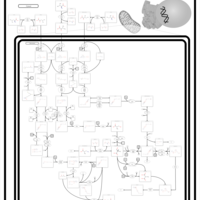 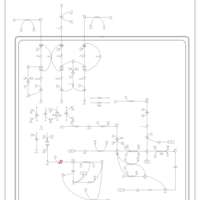 |
| Isovaleric Aciduria |    |
| Saccharopinuria/Hyperlysinemia II | 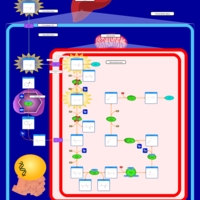  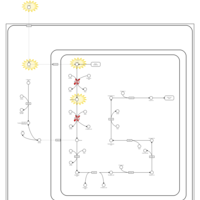 |
| Dimethylglycine Dehydrogenase Deficiency |  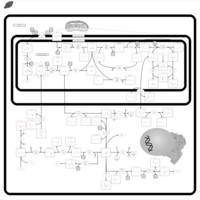  |
| 4-Hydroxybutyric Aciduria/Succinic Semialdehyde Dehydrogenase Deficiency | 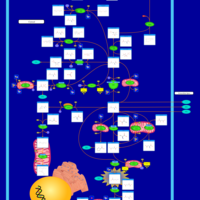 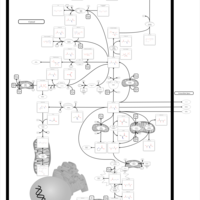 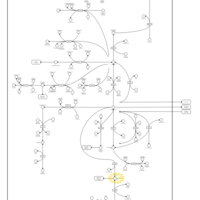 |
| Sarcosinemia |    |
| Lactic Acidemia |  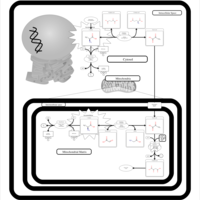 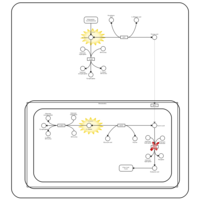 |
| Hyperinsulinism-Hyperammonemia Syndrome |   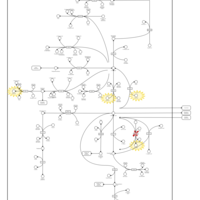 |
| Methylenetetrahydrofolate Reductase Deficiency (MTHFRD) |    |
| Hypermethioninemia |   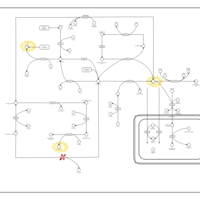 |
| Hereditary Coproporphyria (HCP) | 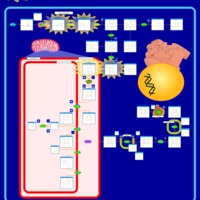 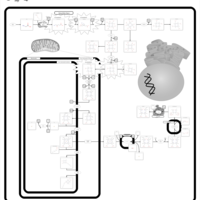  |
| Acute Intermittent Porphyria | 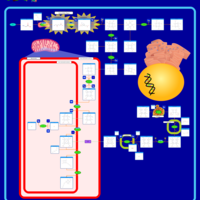 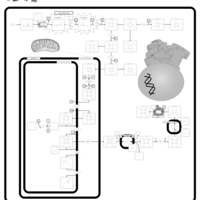 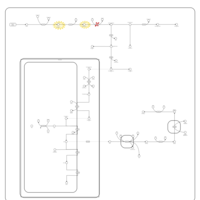 |
| Congenital Erythropoietic Porphyria (CEP) or Gunther Disease |  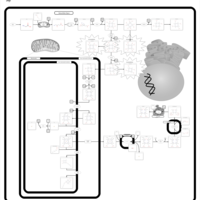 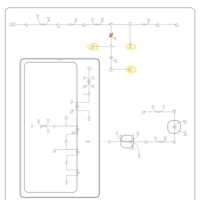 |
| Porphyria Variegata (PV) | 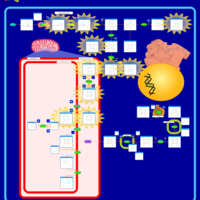 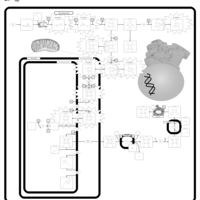 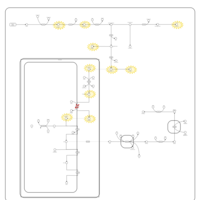 |
| Metachromatic Leukodystrophy (MLD) | 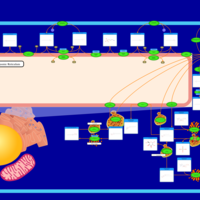 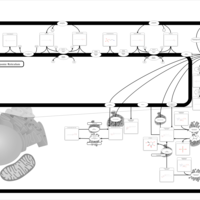  |
| Globoid Cell Leukodystrophy | 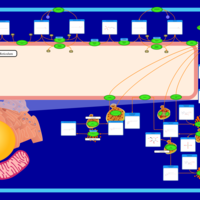 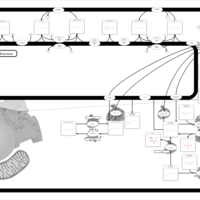 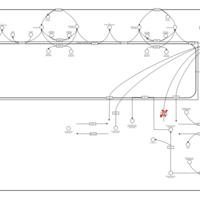 |
| Gaucher Disease |  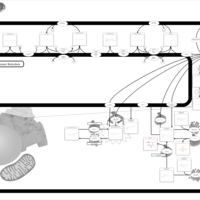 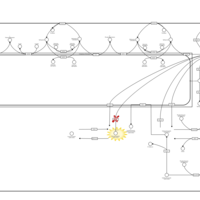 |
| Pyruvate Carboxylase Deficiency | 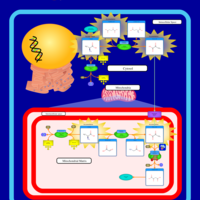 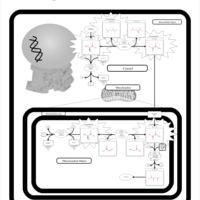 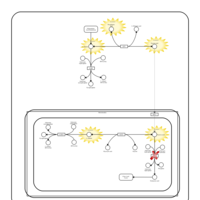 |
| GABA-Transaminase Deficiency | 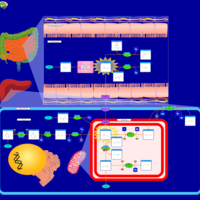 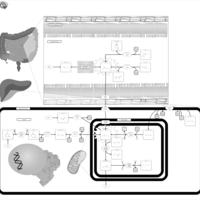 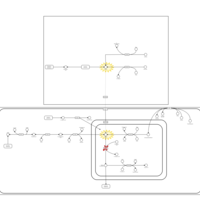 |
| Primary Hyperoxaluria Type I | 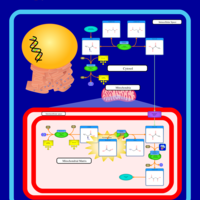 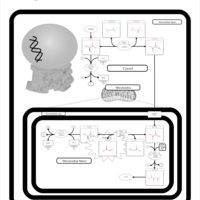 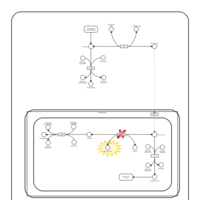 |
| Argininemia | 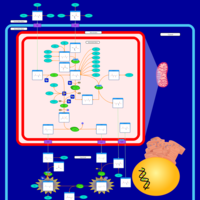  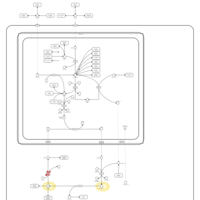 |
| Hyperprolinemia Type II | 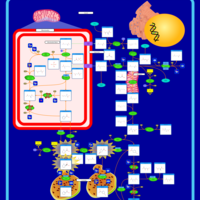 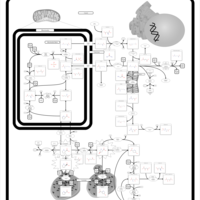 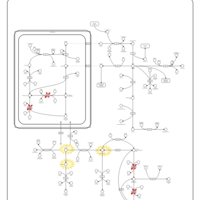 |
| Hyperprolinemia Type I | 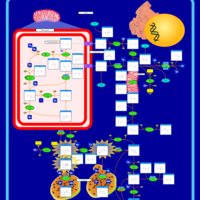 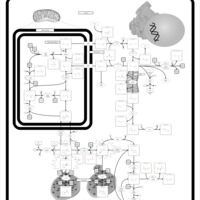 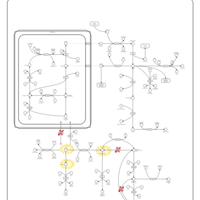 |
| Arginine: Glycine Amidinotransferase Deficiency (AGAT Deficiency) |   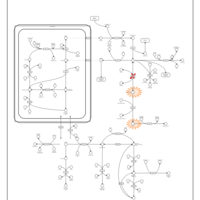 |
| Ornithine Aminotransferase Deficiency (OAT Deficiency) |    |
| Tyrosinemia Type 2 (or Richner-Hanhart syndrome) |   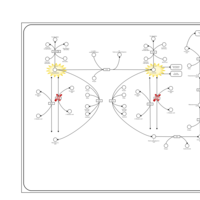 |
| Tyrosinemia Type 3 (TYRO3) | 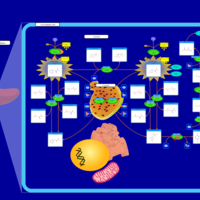   |
| Methylmalonate Semialdehyde Dehydrogenase Deficiency |    |
| Homocarnosinosis | 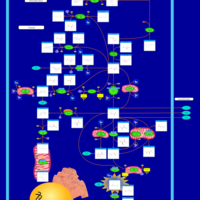   |
| Disulfiram Action Pathway |   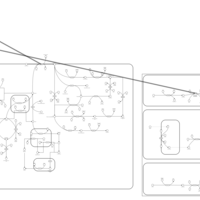 |
| Methotrexate Action Pathway | 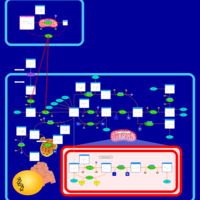 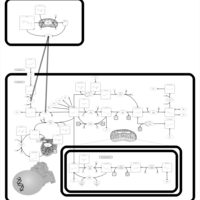 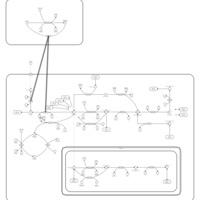 |
| Spermidine and Spermine Biosynthesis | 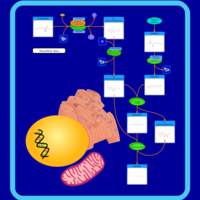 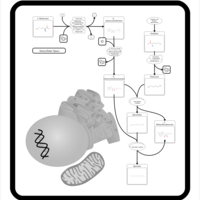  |
| Threonine and 2-Oxobutanoate Degradation | 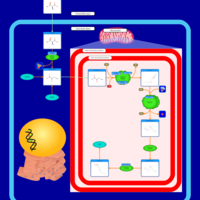 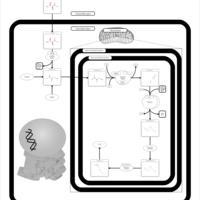  |
| Homocysteine Degradation | 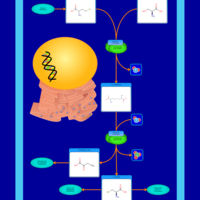 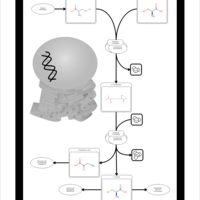  |
| Carnitine Synthesis |    |
| Dimethylglycine Dehydrogenase Deficiency |    |
| Hyperglycinemia, non-ketotic |    |
| Ureidopropionase Deficiency |    |
| Carnosinuria, carnosinemia |  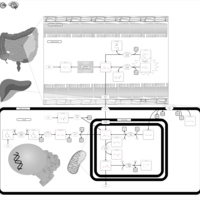 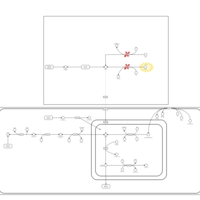 |
| Tyrosinemia, transient, of the newborn |    |
| Tyrosine hydroxylase deficiency | 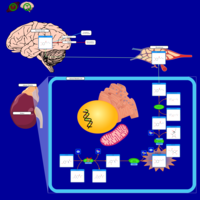 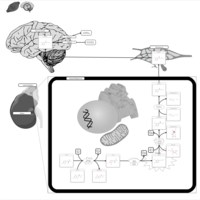  |
| Dopamine beta-hydroxylase deficiency |    |
| Beta-mercaptolactate-cysteine disulfiduria | 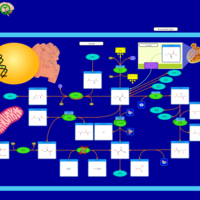 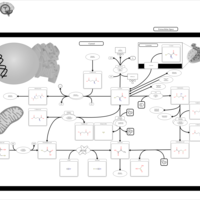 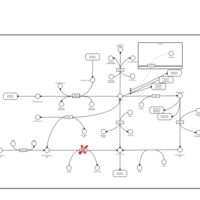 |
| Malonyl-coa decarboxylase deficiency | 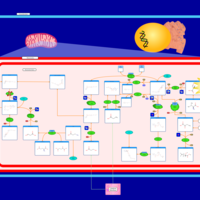 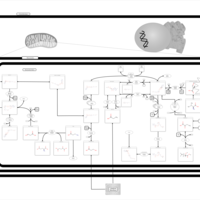 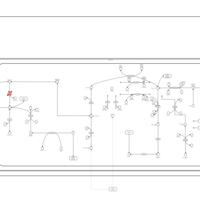 |
| Hypophosphatasia | 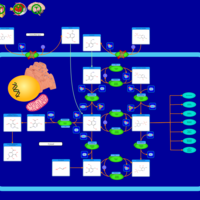 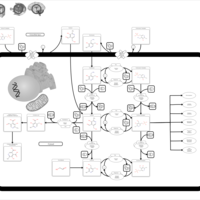 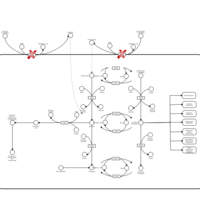 |
| Creatine deficiency, guanidinoacetate methyltransferase deficiency |    |
| Hyperornithinemia with gyrate atrophy (HOGA) |    |
| Hyperornithinemia-hyperammonemia-homocitrullinuria [HHH-syndrome] |    |
| L-arginine:glycine amidinotransferase deficiency |    |
| Gamma-cystathionase deficiency (CTH) | 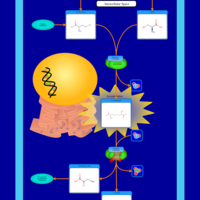 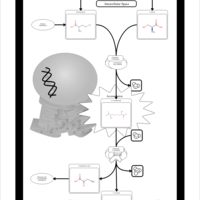 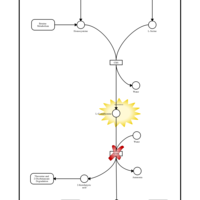 |
| Homocystinuria, cystathionine beta-synthase deficiency | 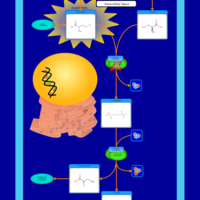   |
| 3-hydroxyisobutyric acid dehydrogenase deficiency |   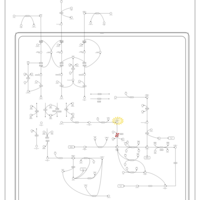 |
| 3-hydroxyisobutyric aciduria |  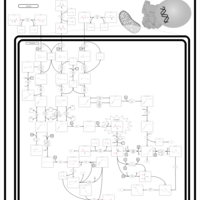 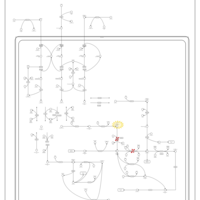 |
| Isobutyryl-coa dehydrogenase deficiency |    |
| Isovaleric acidemia |    |
| Fabry disease |    |
| Krabbe disease |    |
| Hyperlysinemia I, Familial |  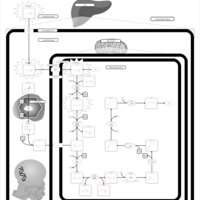 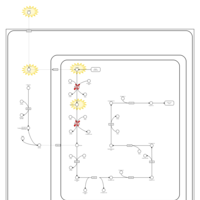 |
| Hyperlysinemia II or Saccharopinuria | 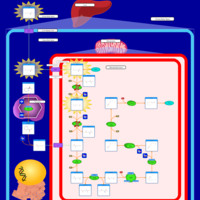 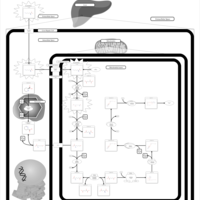  |
| Monoamine oxidase-a deficiency (MAO-A) |    |
| Methylenetetrahydrofolate Reductase Deficiency (MTHFRD) | 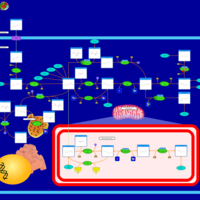 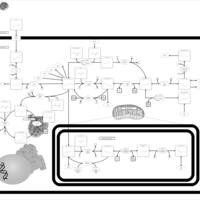 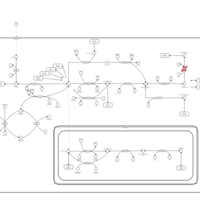 |
| Glycogen synthetase deficiency | 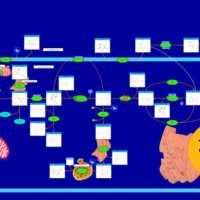 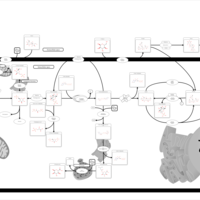  |
| Glycogenosis, Type III. Cori disease, Debrancher glycogenosis | 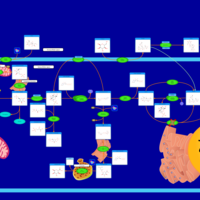 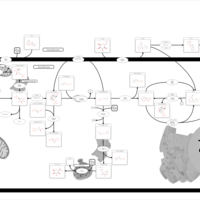 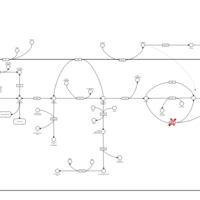 |
| Glycogenosis, Type IV. Amylopectinosis, Anderson disease | 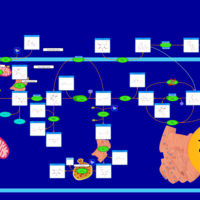 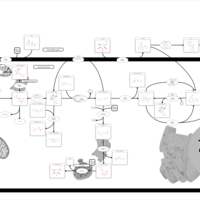 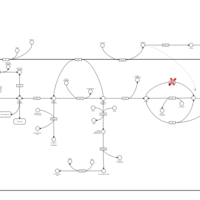 |
| Glycogenosis, Type VI. Hers disease | 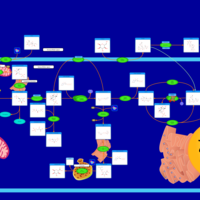 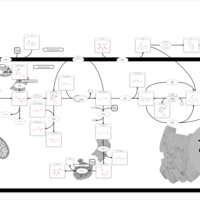 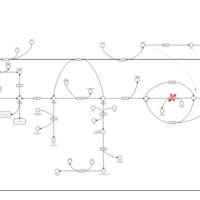 |
| Mucopolysaccharidosis VI. Sly syndrome |    |
| Sucrase-isomaltase deficiency | 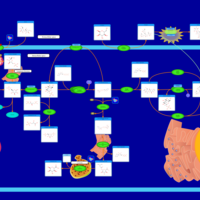 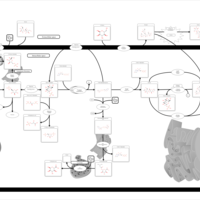 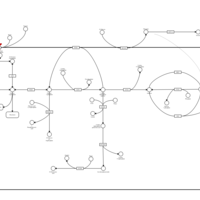 |
| Succinic semialdehyde dehydrogenase deficiency | 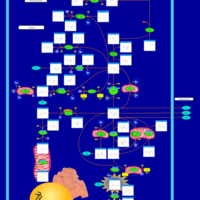 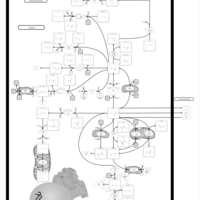 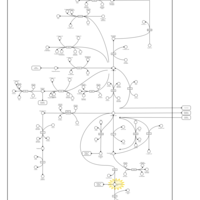 |
| Homocystinuria-megaloblastic anemia due to defect in cobalamin metabolism, cblG complementation type |    |
| Pyridoxine dependency with seizures |  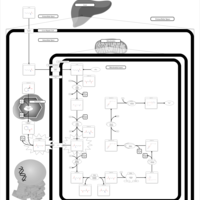 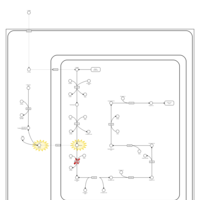 |
| 2-aminoadipic 2-oxoadipic aciduria | 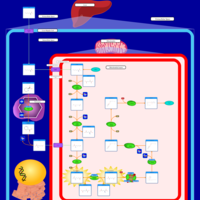  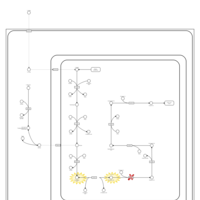 |
| 3-Phosphoglycerate dehydrogenase deficiency | 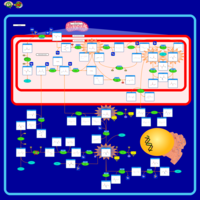 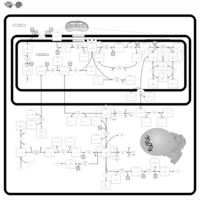 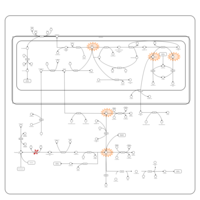 |
| Cystinosis, ocular nonnephropathic | 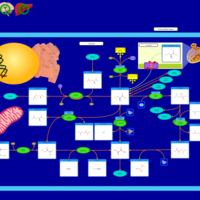 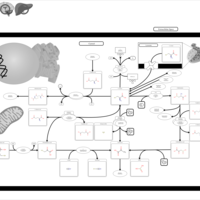 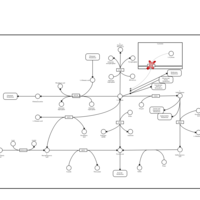 |
| Folate malabsorption, hereditary | 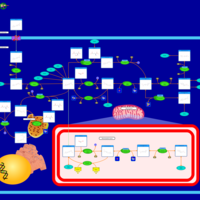 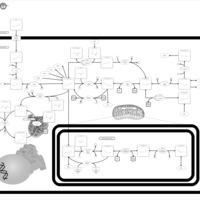 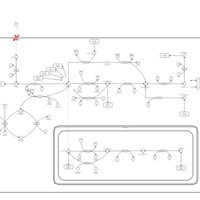 |
| Glutaminolysis and Cancer | 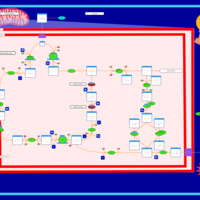 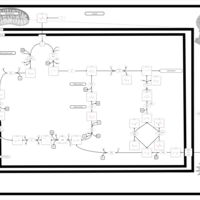 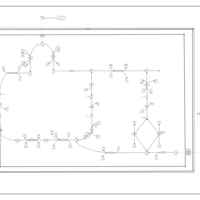 |
| Sarcosine Oncometabolite Pathway |  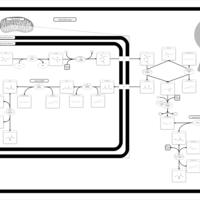  |
